Download as PDF, PPTX








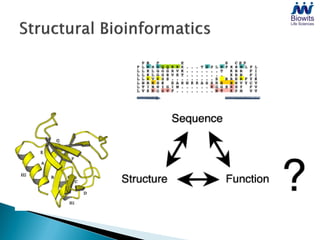






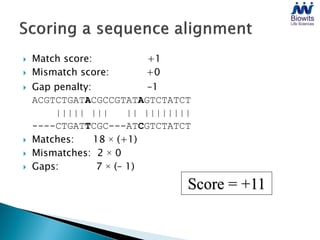










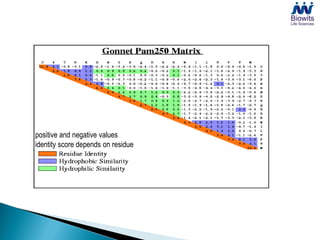





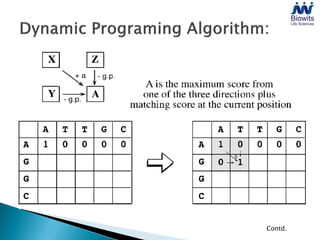
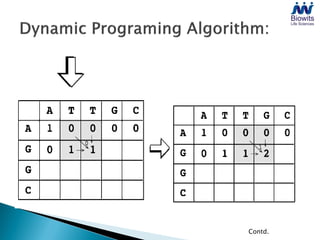

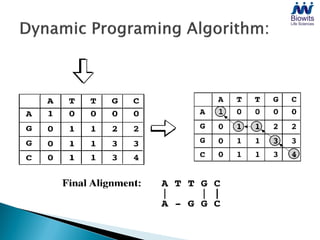



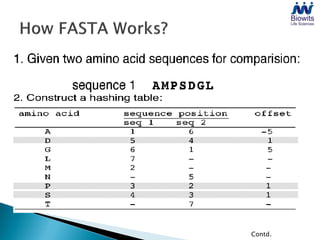


















Bioinformatics emerged from the marriage of computer science and molecular biology to analyze massive amounts of biological data, like that produced by the Human Genome Project. It uses algorithms and techniques from computer science to solve problems in molecular biology, like comparing genomic sequences to understand evolution. As genomic data exploded publicly, bioinformatics was needed to efficiently store, analyze, and make sense of this information, which has applications in molecular medicine, drug development, agriculture, and more.























































































