Downloaded 648 times



















![FASTA Format
• simple format used by almost all programs
• >header line with a [return] at end
• Sequence (no specific requirements for line
length, characters, etc)
>URO1 uro1.seq Length: 2018 November 9, 2000 11:50 Type: N Check: 3854 ..
CGCAGAAAGAGGAGGCGCTTGCCTTCAGCTTGTGGGAAATCCCGAAGATGGCCAAAGACA
ACTCAACTGTTCGTTGCTTCCAGGGCCTGCTGATTTTTGGAAATGTGATTATTGGTTGTT
GCGGCATTGCCCTGACTGCGGAGTGCATCTTCTTTGTATCTGACCAACACAGCCTCTACC
CACTGCTTGAAGCCACCGACAACGATGACATCTATGGGGCTGCCTGGATCGGCATATTTG
TGGGCATCTGCCTCTTCTGCCTGTCTGTTCTAGGCATTGTAGGCATCATGAAGTCCAGCA
GGAAAATTCTTCTGGCGTATTTCATTCTGATGTTTATAGTATATGCCTTTGAAGTGGCAT
CTTGTATCACAGCAGCAACACAACAAGACTTTTTCACACCCAACCTCTTCCTGAAGCAGA
TGCTAGAGAGGTACCAAAACAACAGCCCTCCAAACAATGATGACCAGTGGAAAAACAATG
GAGTCACCAAAACCTGGGACAGGCTCATGCTCCAGGACAATTGCTGTGGCGTAAATGGTC
CATCAGACTGGCAAAAATACACATCTGCCTTCCGGACTGAGAATAATGATGCTGACTATC
CCTGGCCTCGTCAATGCTGTGTTATGAACAATCTTAAAGAACCTCTCAACCTGGAGGCTT 22](https://image.slidesharecdn.com/blastfasta-4-121128073311-phpapp02/85/Blast-fasta-4-22-320.jpg)






![BLAST Searches GenBank
[BLAST= Basic Local Alignment Search Tool]
The NCBI BLAST web server lets you compare your
query sequence to various sections of GenBank:
– nr = non-redundant (main sections)
– month = new sequences from the past few weeks
– refseq_rna
– RNA entries from NCBI's Reference Sequence project
– refseq_genomic
– Genomic entries from NCBI's Reference Sequence project
– ESTs
– Taxon = e.g., human, Drososphila, yeast, E. coli
– proteins (by automatic translation)
– pdb = Sequences derived from the 3-dimensional structure
from Brookhaven Protein Data Bank
29](https://image.slidesharecdn.com/blastfasta-4-121128073311-phpapp02/85/Blast-fasta-4-29-320.jpg)

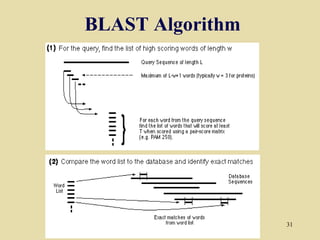
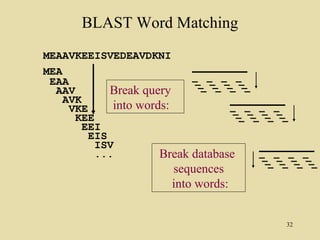

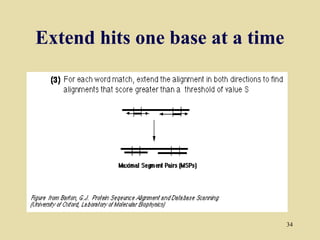






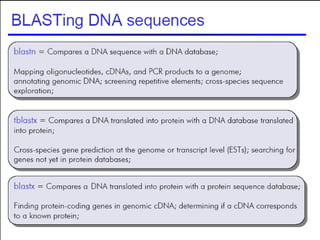




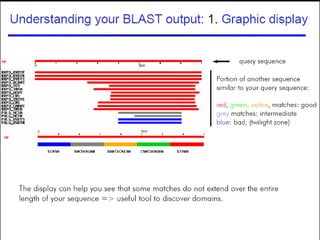














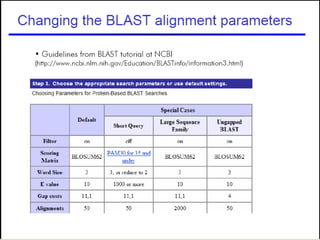



BLAST and FASTA are algorithms for searching sequence databases to find local alignments between a query sequence and database sequences, with BLAST providing faster searches and improved statistical analysis compared to FASTA. Both algorithms work by first identifying short exact matches between sequences and then extending these matches to identify longer regions of similarity. The algorithms model DNA and protein sequence alignments as coin tosses to determine the expected length of the longest matching region between random sequences.



































































