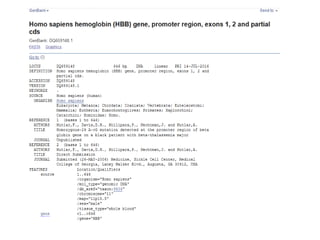
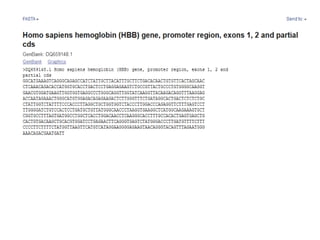
1) The document discusses various bioinformatics databases including nucleotide databases like GenBank that contain nucleic acid sequences, protein databases like PDB that contain 3D protein structures, and specialized databases like dbSNP that contain human single nucleotide variations.
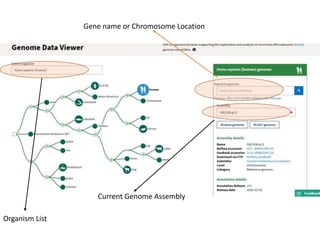
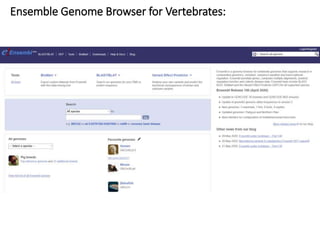
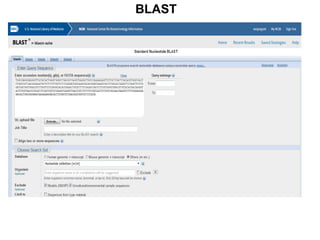
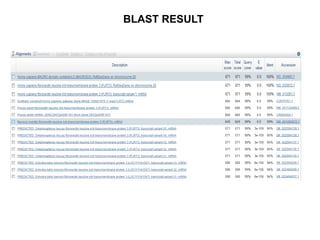
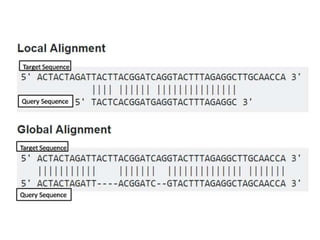
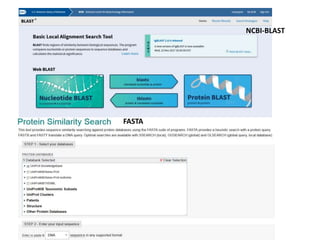
2) It also discusses tools for analyzing sequences like BLAST for similarity searches, multiple sequence alignments, and genome browsers for interactively viewing complete genomes.
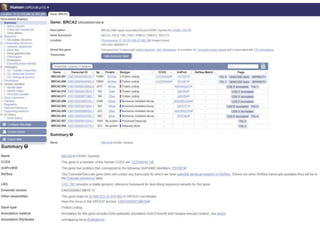
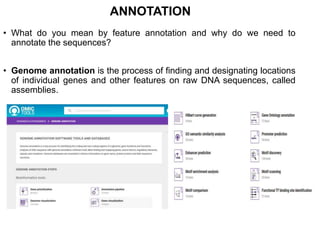
3) Feature annotation is described as the process of identifying genes and other biological features in DNA sequences to increase their usefulness to the scientific community.