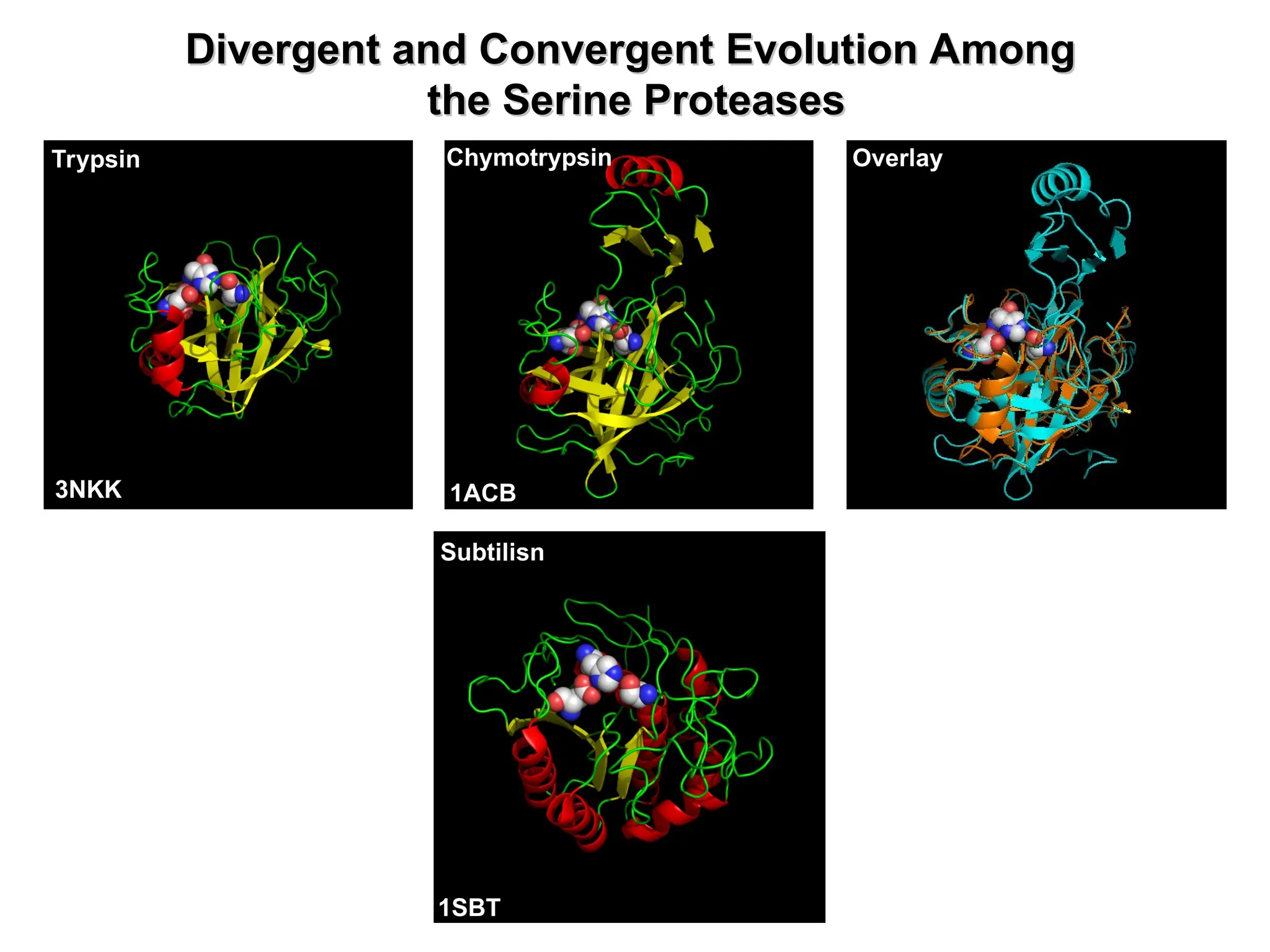
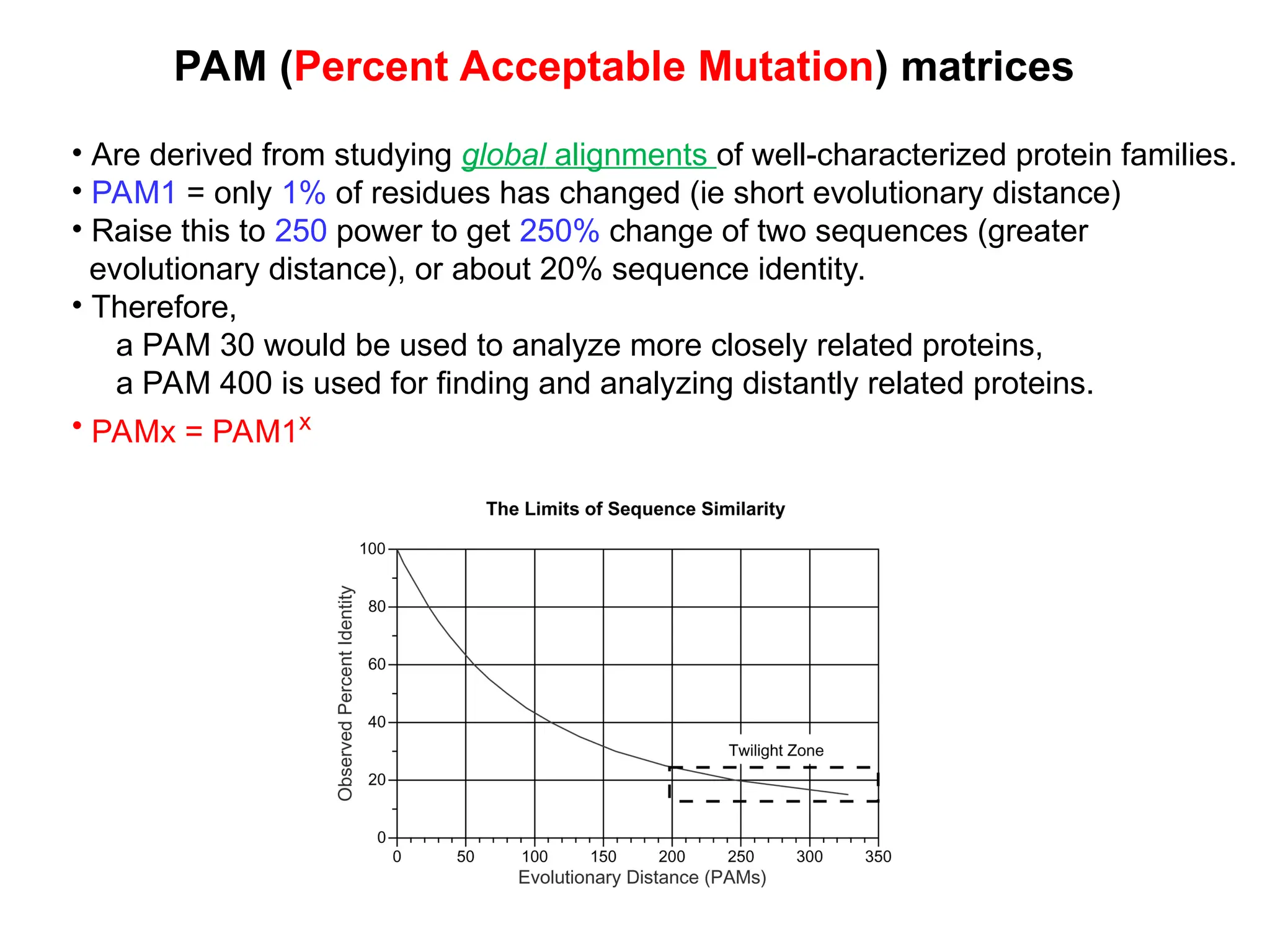
The document discusses protein evolution, including the concepts of homologs, orthologs, and paralogs, and explains the mechanisms such as mutation, recombination, and gene duplication that contribute to genetic diversity. It also covers methods for sequence analysis, emphasizing the importance of sequence alignment to infer functional and structural relationships among genes and proteins, and describes scoring matrices for these analyses. Furthermore, it lists useful bioinformatics resources for protein structure and interaction data.








![Useful Bioinformatics Sites
Useful Bioinformatics Sites
National Center for Biotechnology Information (NCBI)- National Institutes of
Health sponsored sites with rich array of resources and data bases.
[http://www.ncbi.nlm.nih.gov/pubmed]
ExPASy (Swiss Institute of Bioinformatics)- Large number of different
tools for sequence and function analysis. [http://www.expasy.org/tools/]
RCSB Protein Data Bank- Largest data base for curated of protein structures.
[http://www.rcsb.org/pdb/home/home.do]
BioGRID- Large data base of curated protein interaction datasets.
[http://thebiogrid.org/]
Osprey- Software and interactome analysis tools for visualizing interaction
data sets. [http://en.bio-soft.net/protein/Osprey.html]
Tree of Life website- Database information on phylogenetic relationships
among organisms with useful link outs. [http://tolweb.org/tree/]](https://image.slidesharecdn.com/proteinevolutionandsequenceanalysis-250208092639-bcdcad8b/75/Protein-Evolution-and-Sequence-Analysis-ppt-11-2048.jpg)


































![Conservation of Mech Energy [Autosaved].ppt](https://cdn.slidesharecdn.com/ss_thumbnails/conservationofmechenergyautosaved-251110121538-36a25d24-thumbnail.jpg?width=640&height=640&fit=bounds)
































