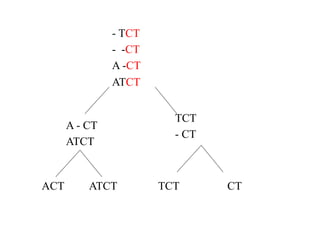
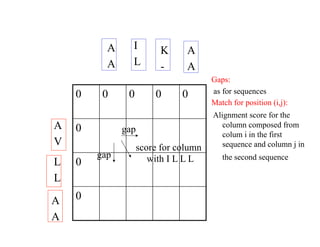
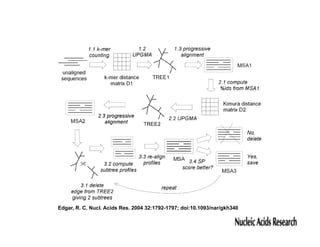
This document discusses multiple sequence alignment. It begins by explaining that pairwise sequence alignment is not reliable for more distantly related sequences, as there may be many possible alignments with the same score. Multiple sequence alignment allows discovering conserved motifs across a protein family. The document then discusses different scoring systems for multiple sequence alignments, including sum-of-pairs and entropy-based scores. It also describes the dynamic programming solution and progressive alignment approaches like CLUSTALW and T-COFFEE. The document concludes by mentioning faster methods like MUSCLE that use hashing to find short matches and build an initial sequence similarity tree.