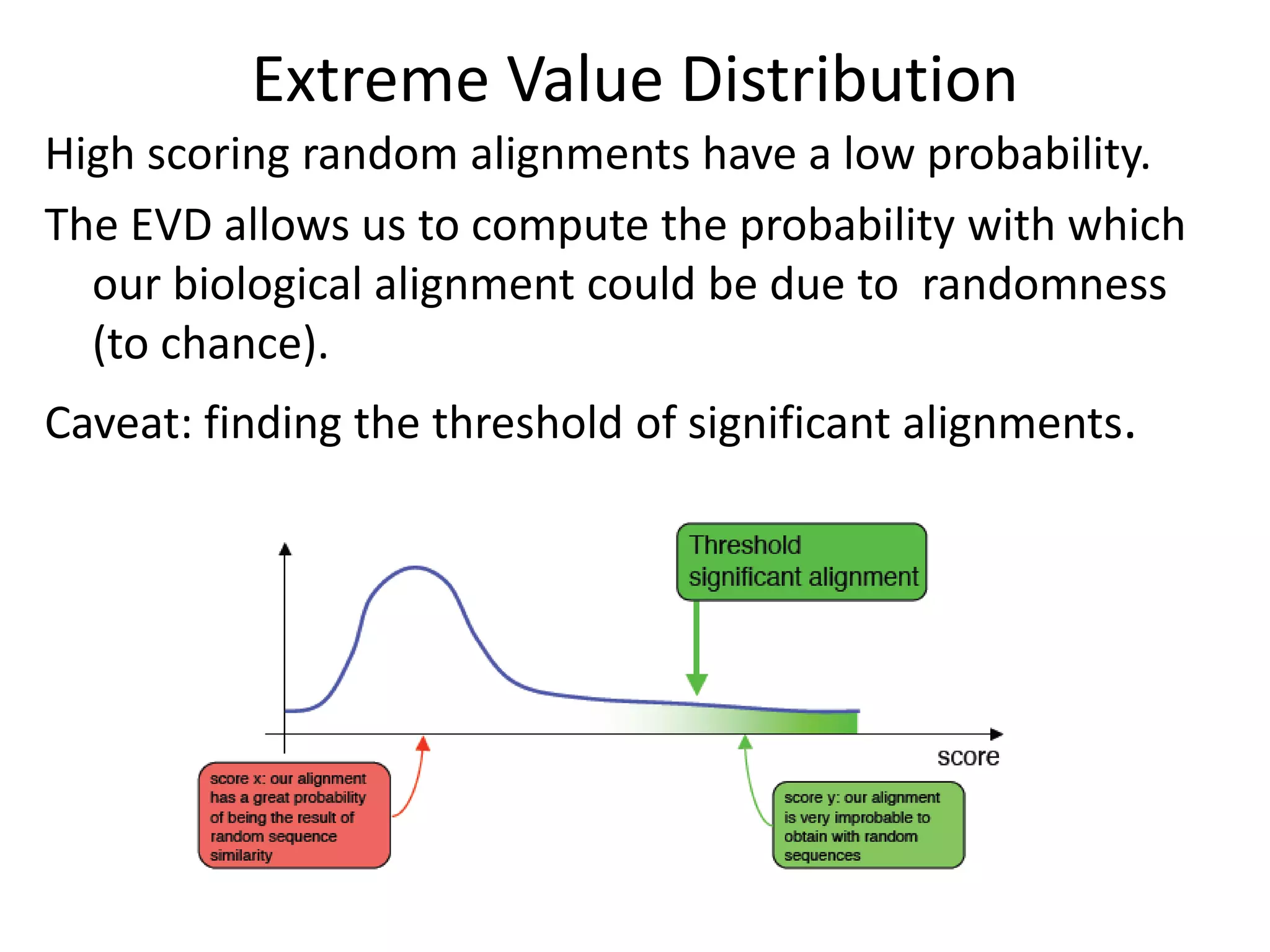
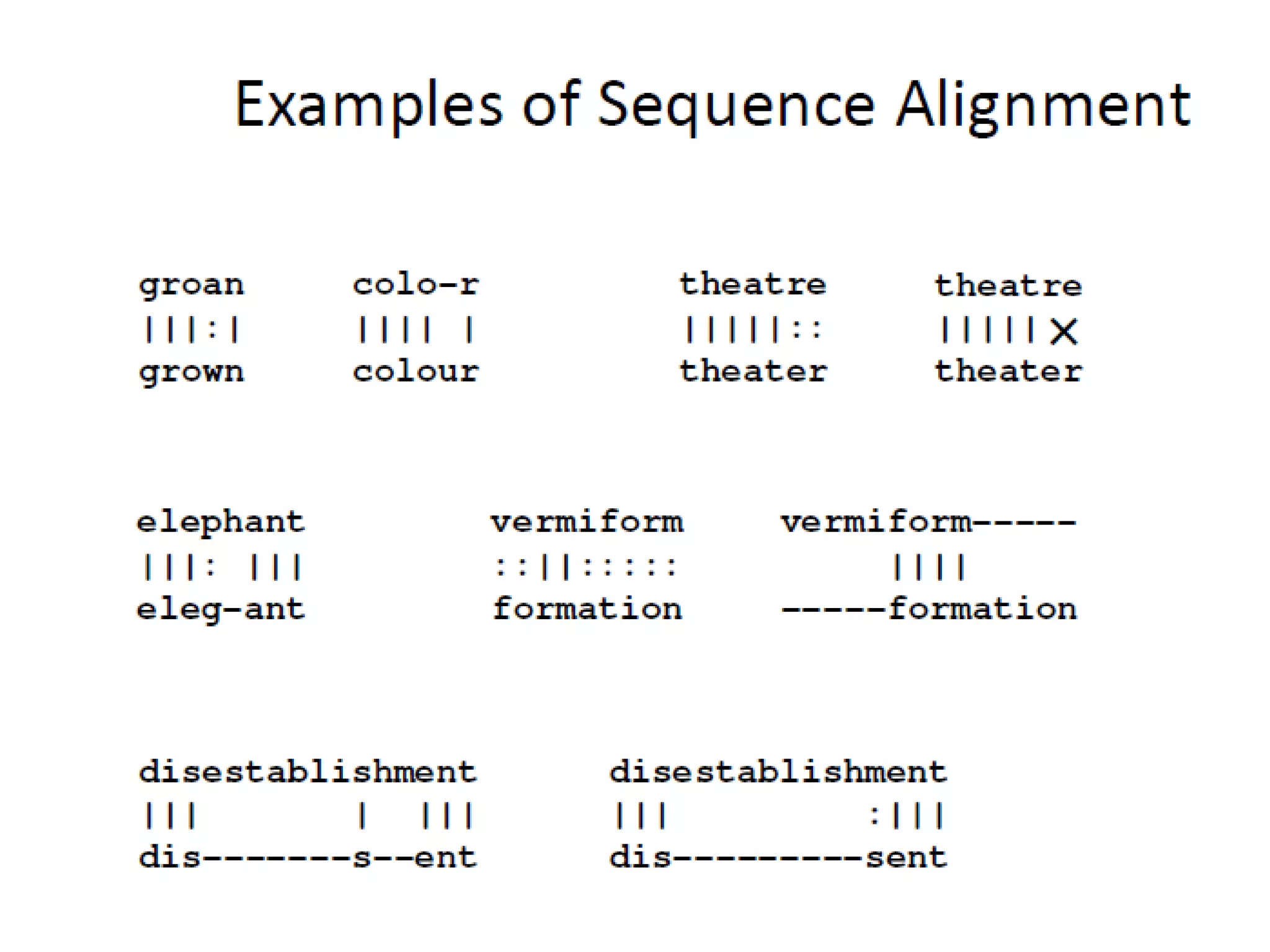
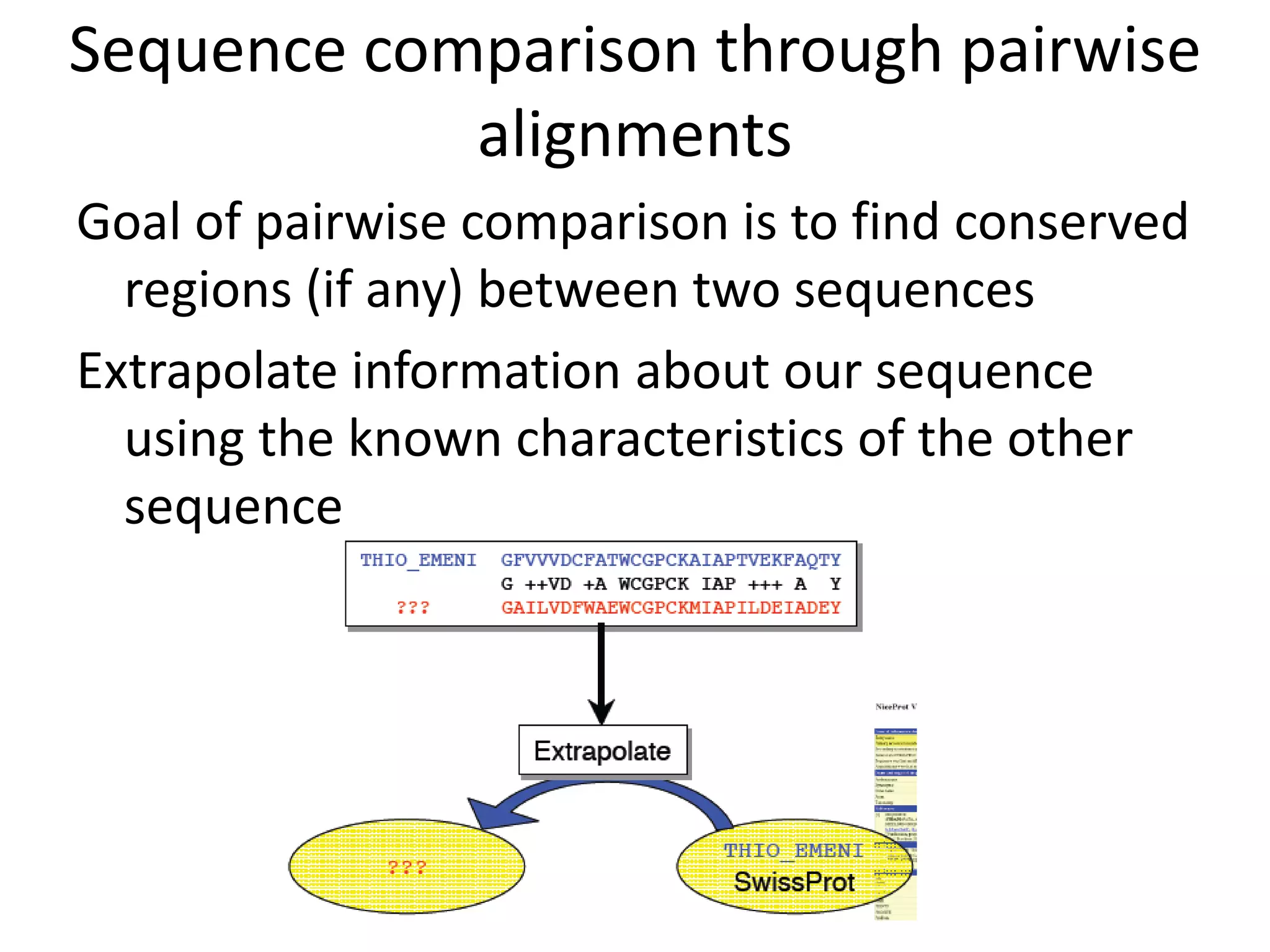
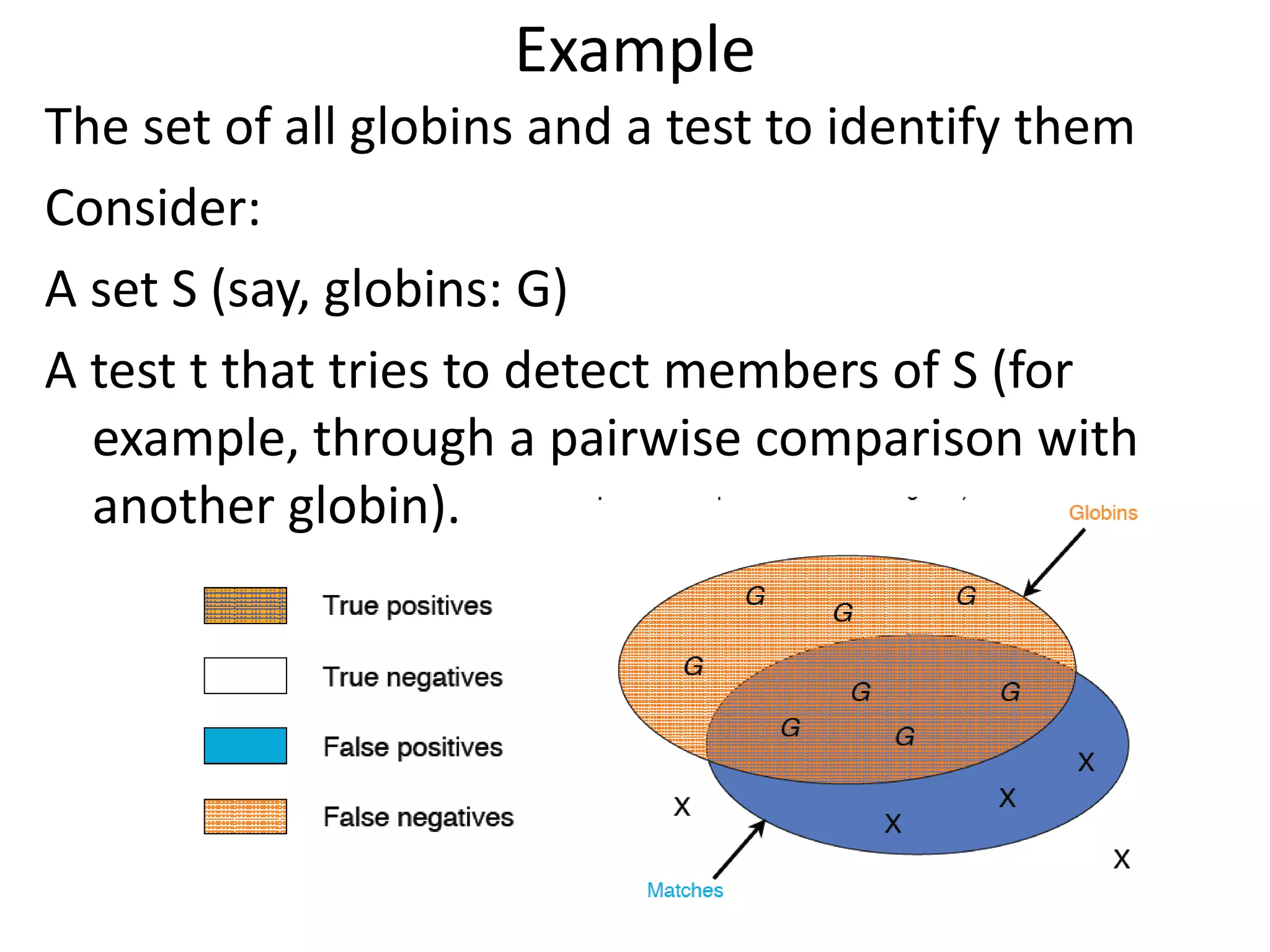
The document discusses pairwise sequence alignment, which involves mapping residues between two sequences to find conserved regions and maximize similarity score. It describes how alignment is used to infer homology and related applications. Key concepts covered include substitution matrices like PAM and BLOSUM that account for amino acid substitutability, and dot plots which provide a graphical representation of sequence similarity.






![Important questions
Q. What do we want to align and how?
A: Two sequences (nucleotide or protein) through pairwise
alignment
Or To find similar sequences in a database against our query
sequence by multiple sequence alignment
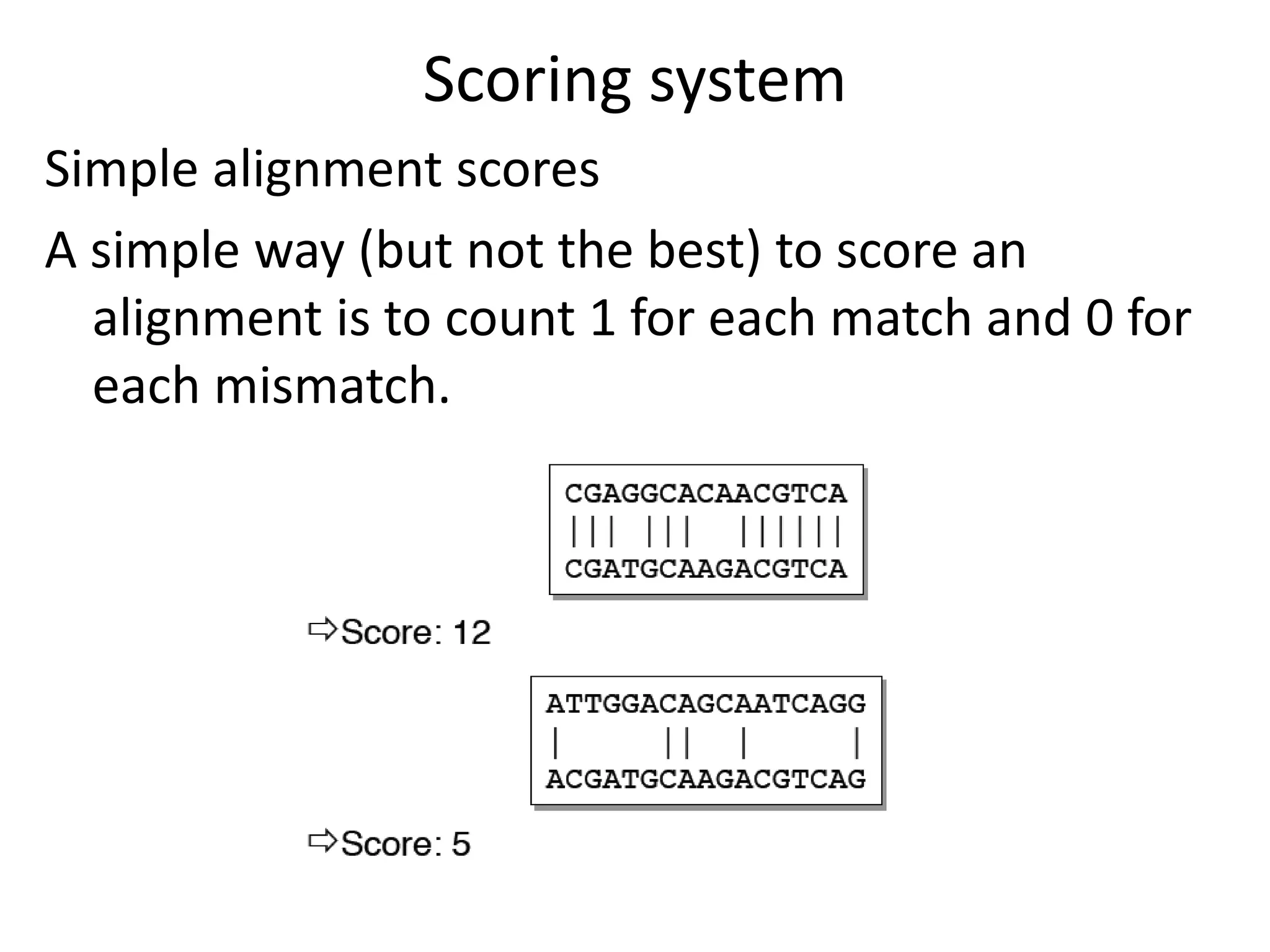
Q. How do we “score” an alignment?
Simple scoring (match= 1, mismatch= 0),
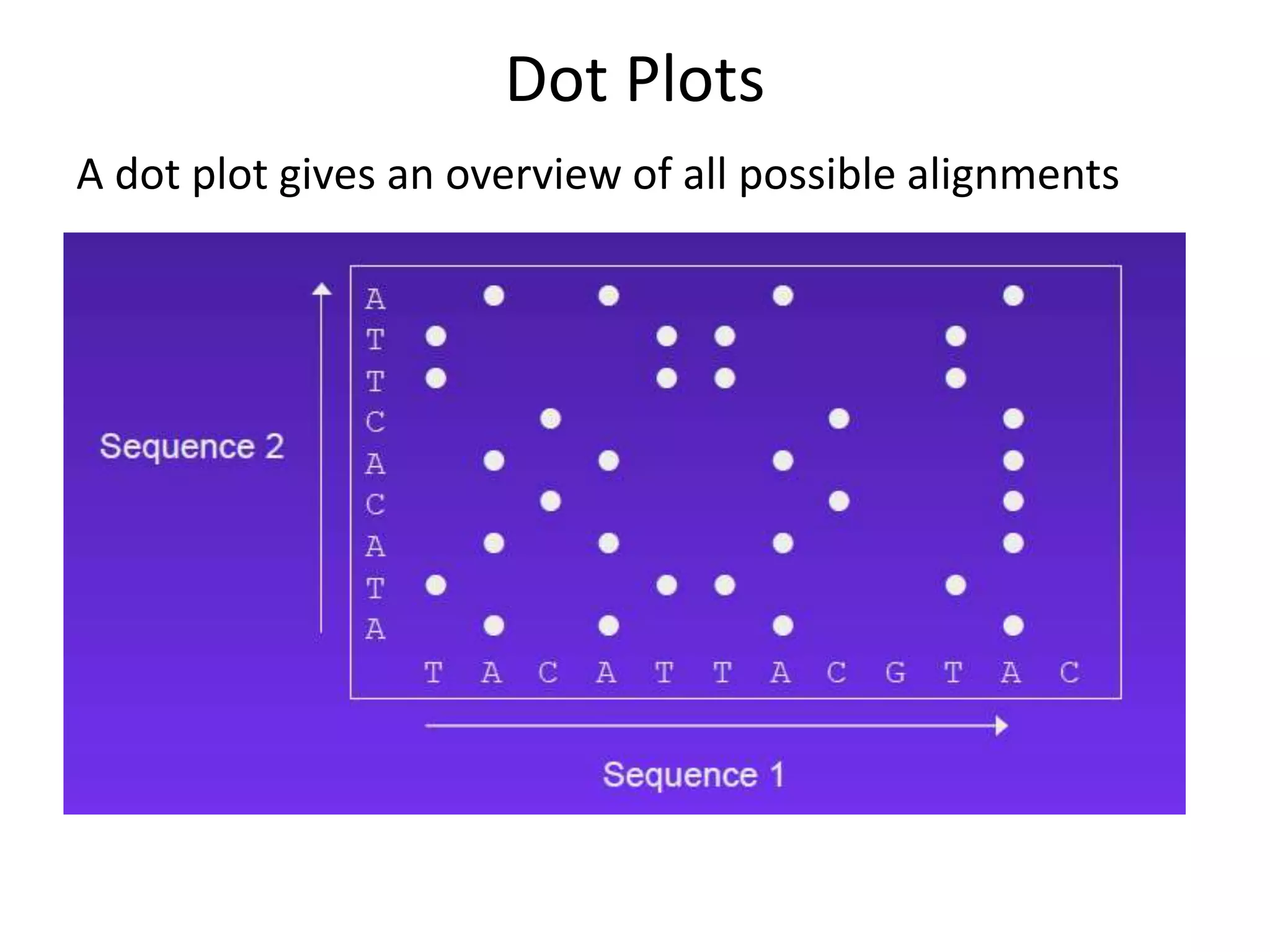
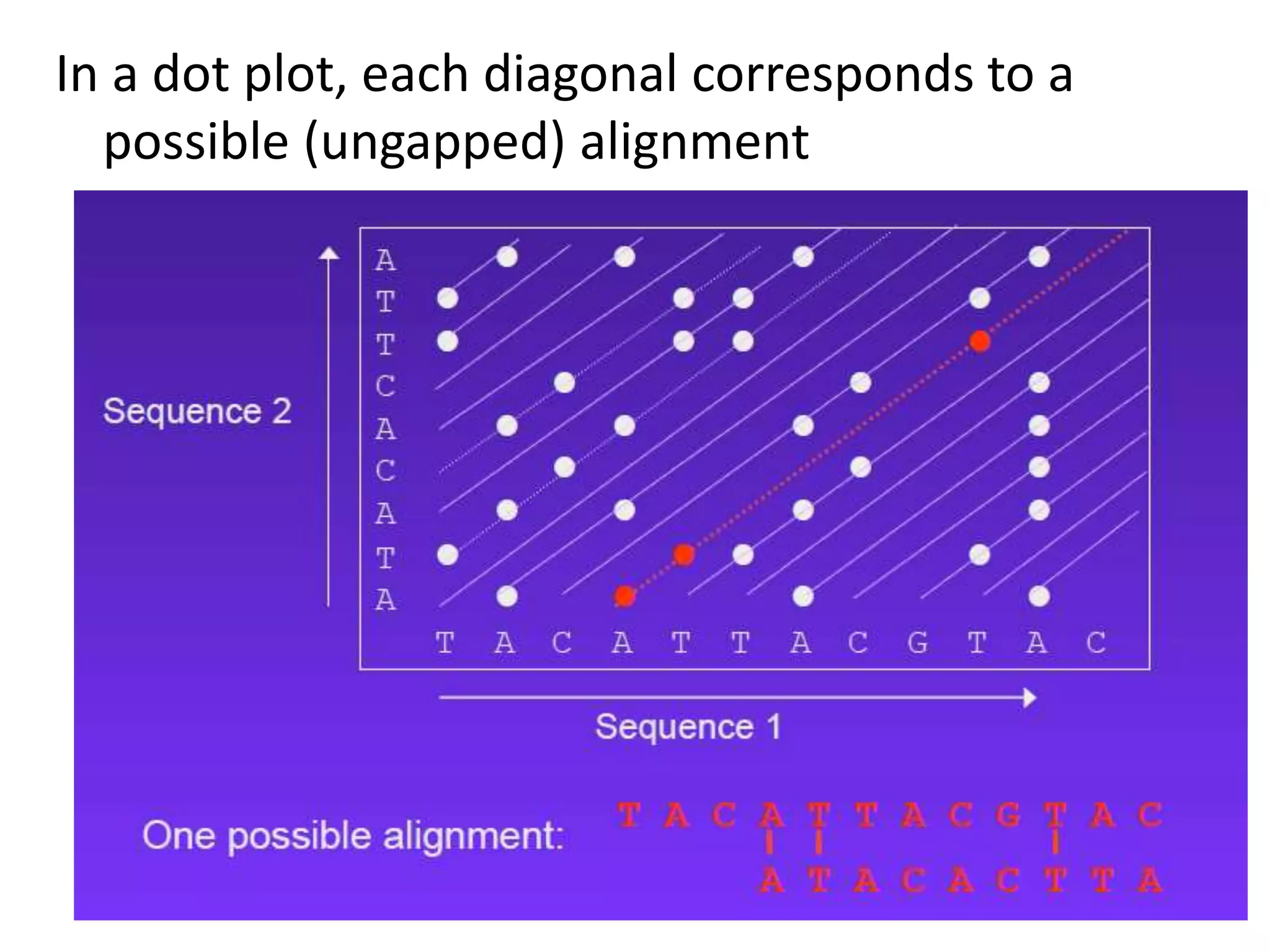
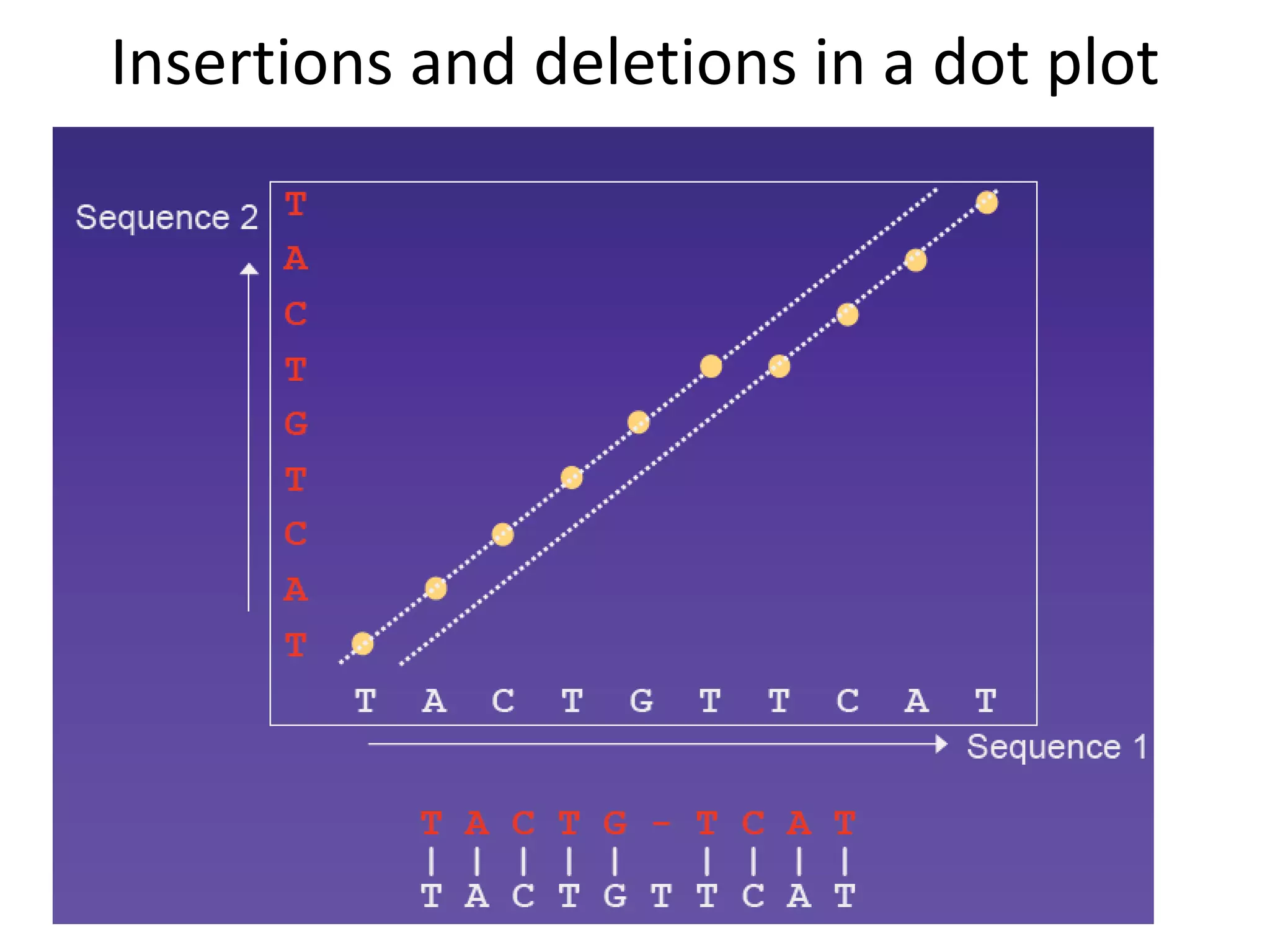
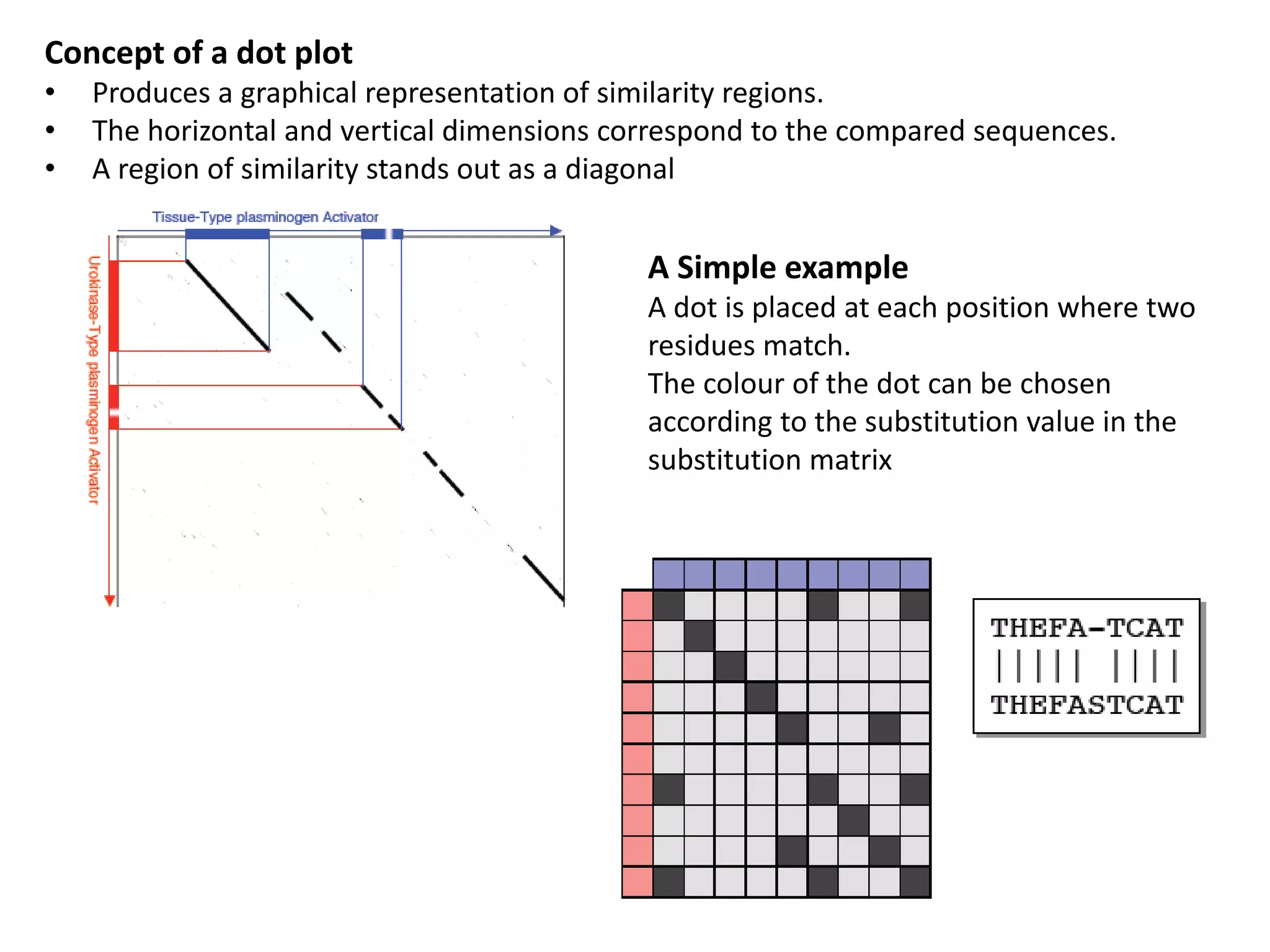
Dot plots (graphical representation)
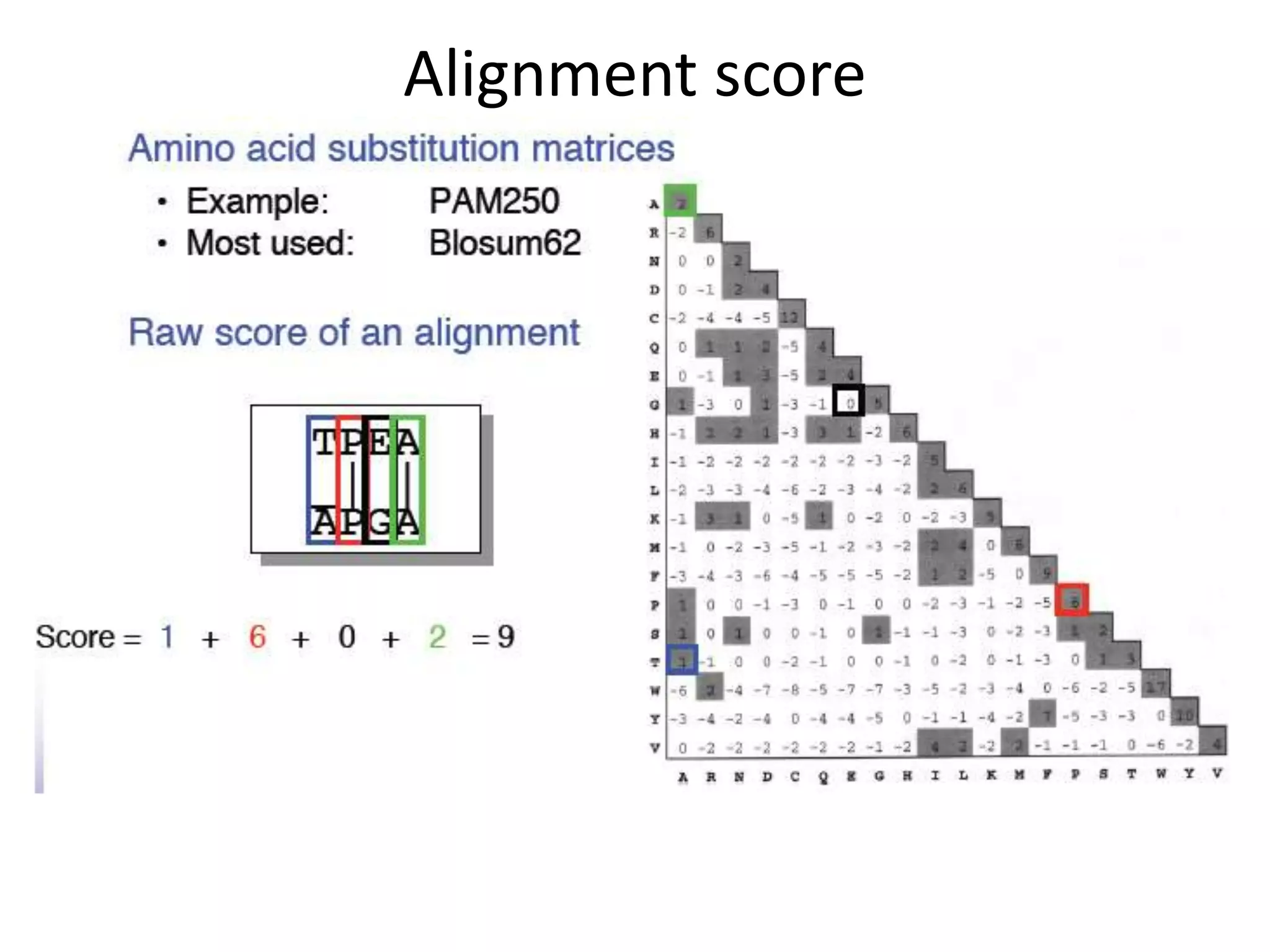
Substitution matrices (PAM and BLOSUM)[s(a,b)
indicates score of aligning character a with character b;
Also accounts for relative substitutability of amino acid
pairs in the context of evolution]
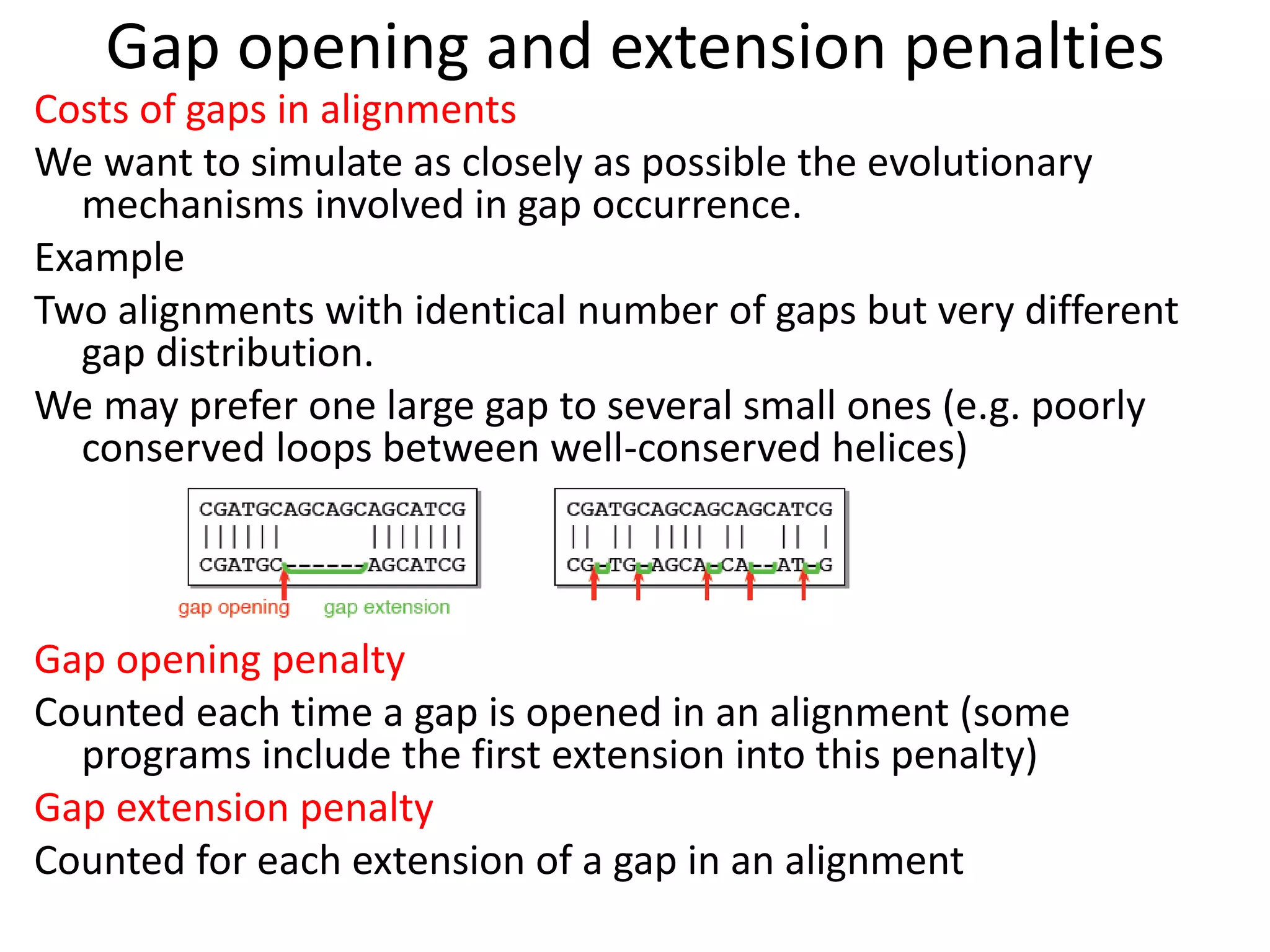
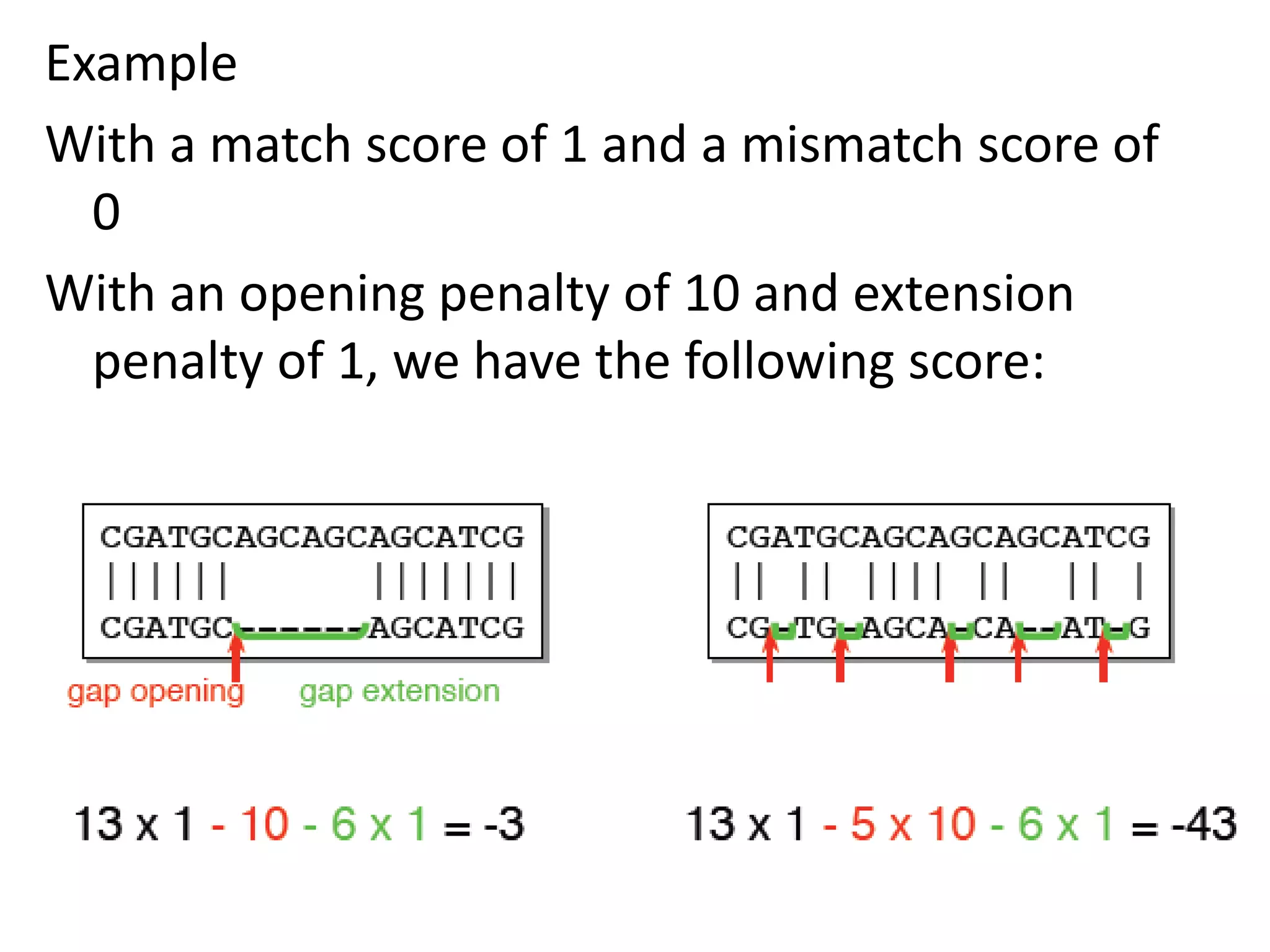
Gap penalty function: w(k) indicates cost of a gap of
length k](https://image.slidesharecdn.com/4-230521122917-4edd4289/75/4-sequence-alignment-pptx-8-2048.jpg)


![Evolution of sequences
Sequences evolve through mutation and selection
[Selective pressure is different for each residue
position in a protein (i.e. conservation of active
site, structure, charge,etc.)]
Modular nature of proteins [Nature keeps re-using
domains]
Alignments try to tell the evolutionary story of the
proteins
Relationships](https://image.slidesharecdn.com/4-230521122917-4edd4289/75/4-sequence-alignment-pptx-12-2048.jpg)








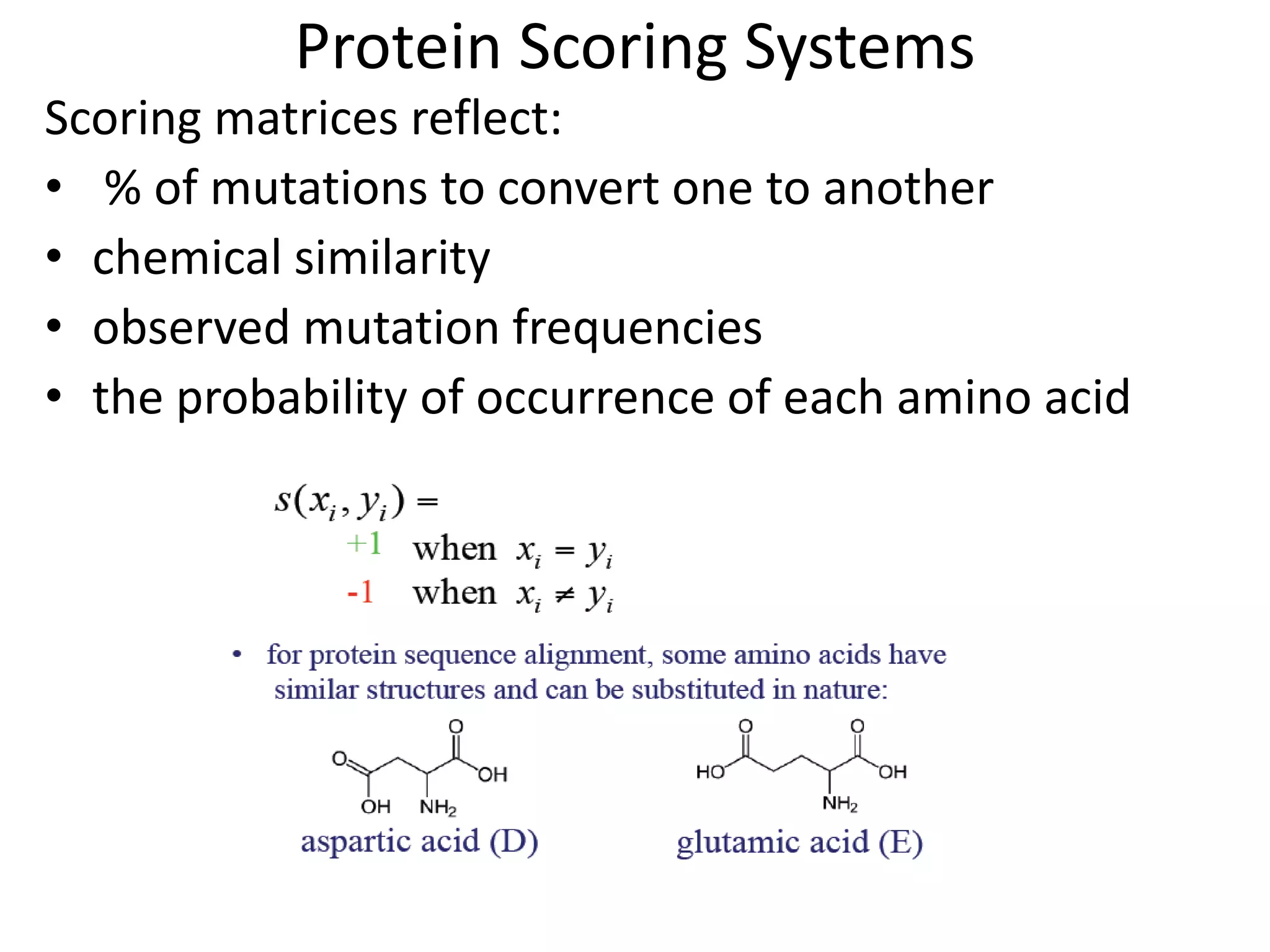
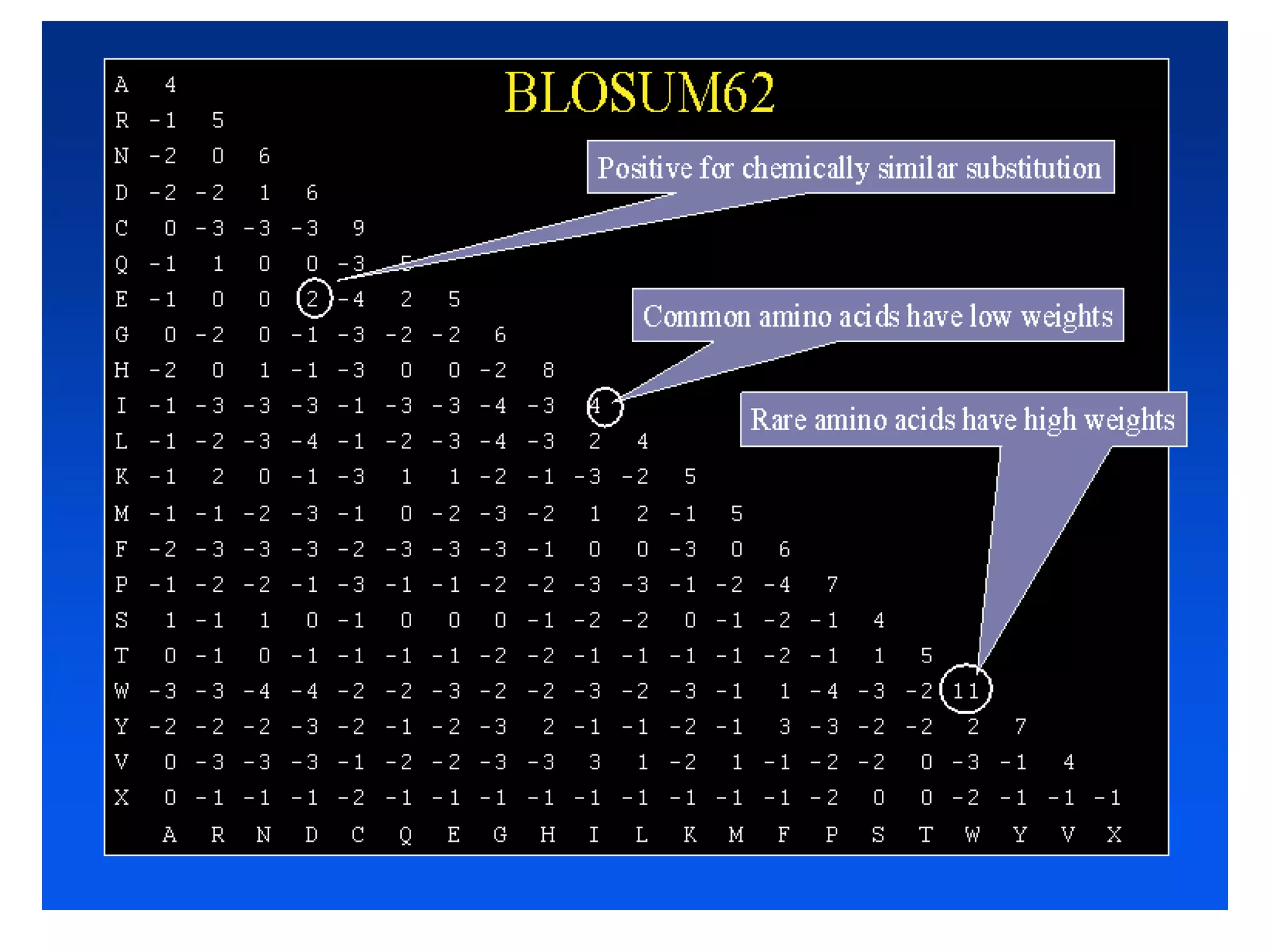
![Substitution Matrices (Log odds matrices)
Two popular sets of matrices for protein
sequences
PAM matrices [Dayhoff et al., 1978]
BLOSUM matrices [Henikoff & Henikoff, 1992]
Both try to capture the relative substitutability of
amino acid pairs in the context of evolution](https://image.slidesharecdn.com/4-230521122917-4edd4289/75/4-sequence-alignment-pptx-28-2048.jpg)