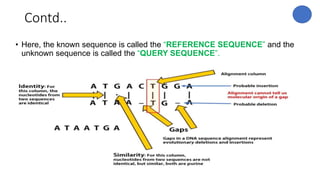
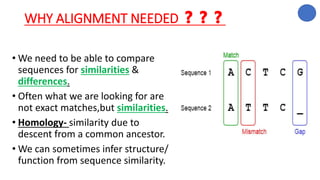
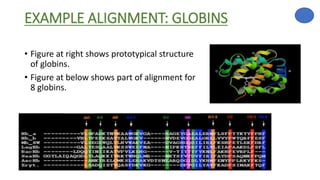
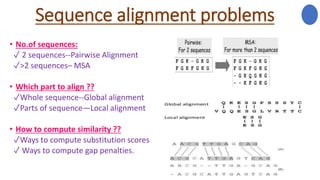
The document discusses sequence alignment, a method used to arrange DNA, RNA, or protein sequences to identify similarities that indicate functional, structural, and evolutionary relationships. It describes the principles, goals, types (pairwise and multiple), tools for alignment, and various issues related to this process, emphasizing the importance of identifying conserved regions and differences. Additionally, it highlights the advantages of sequence alignment in comparing sequences of different lengths and detecting functional orthology.