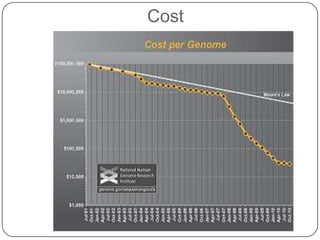
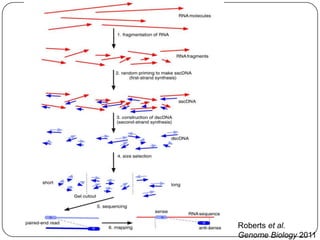
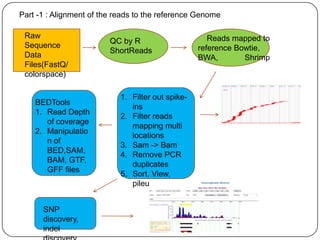
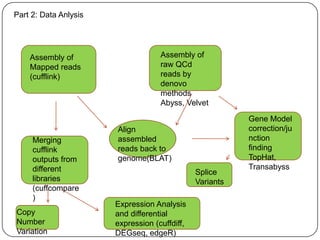
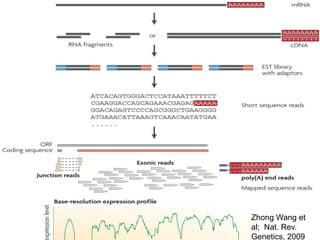
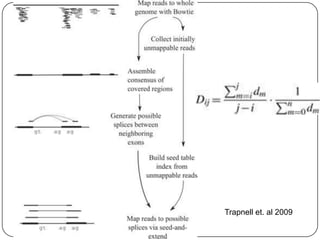
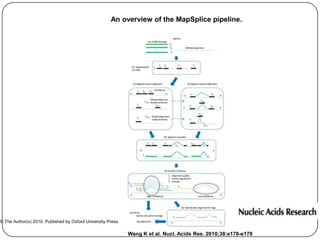
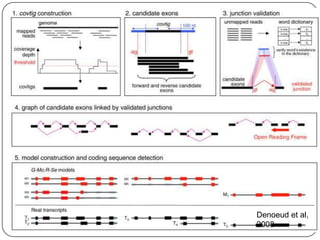
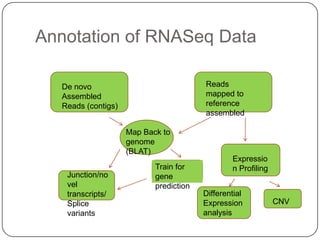
The document discusses next generation sequencing methods and RNA sequencing. It covers topics like sequencing formats, data analysis workflows including mapping, clustering, assembly programs, finding new genes and correcting existing ones. It discusses input file types, calculating sequencing depth, available tools for alignment, output file formats, assembly programs, splice junction prediction, and applications of RNA sequencing like gene expression analysis and annotation.