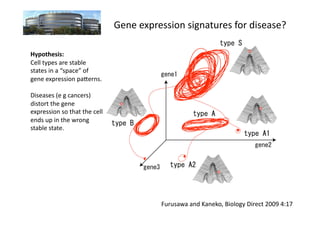
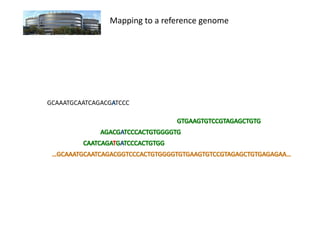
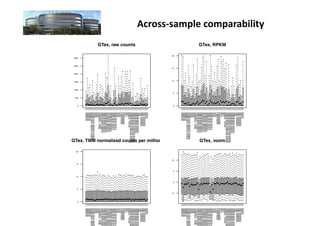
The document discusses RNA-seq analysis. It begins with an introduction to Mikael Huss, a bioinformatics scientist, and provides an overview of how genomics, RNA profiles, protein profiles, and interactomics relate within systems biology. The document then discusses how gene expression analysis can provide insights into basic research questions regarding tissue and cell identity, as well as insights into diseases by identifying genes that are over- or under-expressed in patients. Finally, it provides a brief overview of the typical workflow for RNA-seq analysis, which involves mapping RNA sequencing reads to a reference genome or transcriptome.