Downloaded 147 times













![Bustard output Two tab-separated text files, one row per cluster “ seq.txt” Lane and tile index, x and y coordinates Called sequence as string of A, G, C, T “ prb.txt” Phred-like scores [-40,40] One value per called base](https://image.slidesharecdn.com/20100516bioinformaticskapusheskylecture08-100817174624-phpapp02/85/20100516-bioinformatics-kapushesky_lecture08-16-320.jpg)
![FASTQ format @HWI-EAS225:3:1:2:854#0/1 GGGGGGAAGTCGGCAAAATAGATCCGTAACTTCGGG +HWI-EAS225:3:1:2:854#0/1 a`abbbbabaabbababb^`[aaa`_N]b^ab^``a @HWI-EAS225:3:1:2:1595#0/1 GGGAAGATCTCAAAAACAGAAGTAAAACATCGAACG +HWI-EAS225:3:1:2:1595#0/1 a`abbbababbbabbbbbbabb`aaababab\aa_`](https://image.slidesharecdn.com/20100516bioinformaticskapusheskylecture08-100817174624-phpapp02/85/20100516-bioinformatics-kapushesky_lecture08-17-320.jpg)










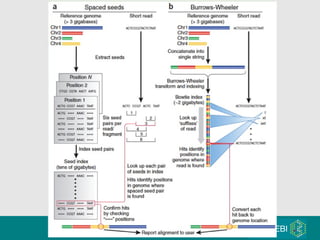





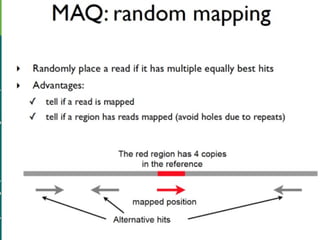
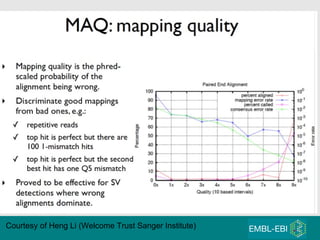

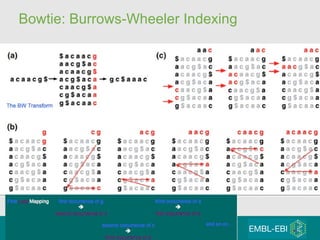






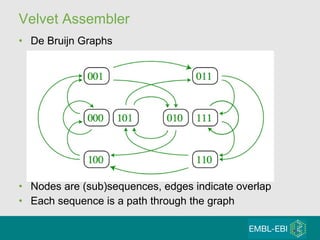

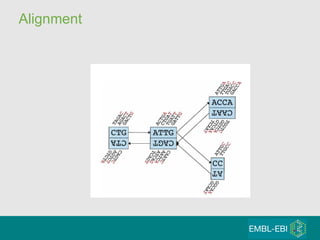


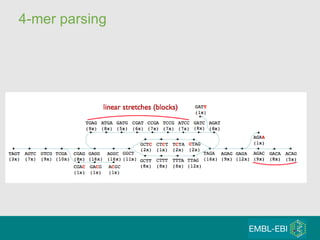
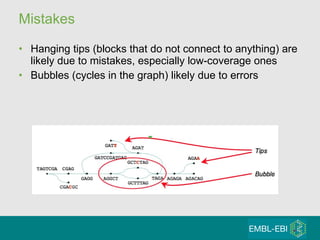
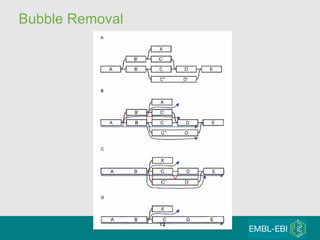







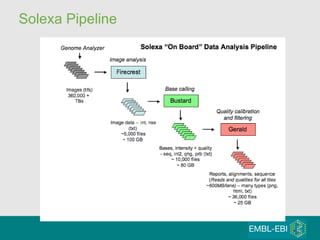
High-throughput sequencing has key differences from Sanger sequencing such as fragments being sequenced in parallel rather than by cloning. Several platforms are discussed including their read lengths, throughput, error rates, and costs. Pair-end and targeted sequencing are also covered. Challenges in bioinformatics include assembly, alignment amid repeats and errors, and downstream analysis tasks. Popular aligners like BWA and Bowtie that use the Burrows-Wheeler transform are fast and accurate. De novo assembly requires specialized tools to handle short reads. RNA-seq has additional complexities in assembly.


































































