Downloaded 64 times



![Align (using BWA)
1) Index the potato genome assembly
bwa index [-a bwtsw|div|is] [-c]
<in.fasta>
2) Perform the alignment
bwa aln [options] <in.fasta>
<in.fq>
3) Output results in SAM format (single end)
bwa samse <in.fasta> <in.sai>
<in.fq> 5](https://image.slidesharecdn.com/pipeline-03x-130220181153-phpapp02/75/Creating-a-SNP-calling-pipeline-5-2048.jpg)
![Align (using Bowtie)
1) Index the potato genome assembly
bowtie-build [options] <in.fasta>
<ebwt>
2) Perform the alignment and output results
bowtie [options] <ebwt> <in.fq>](https://image.slidesharecdn.com/pipeline-03x-130220181153-phpapp02/75/Creating-a-SNP-calling-pipeline-6-2048.jpg)
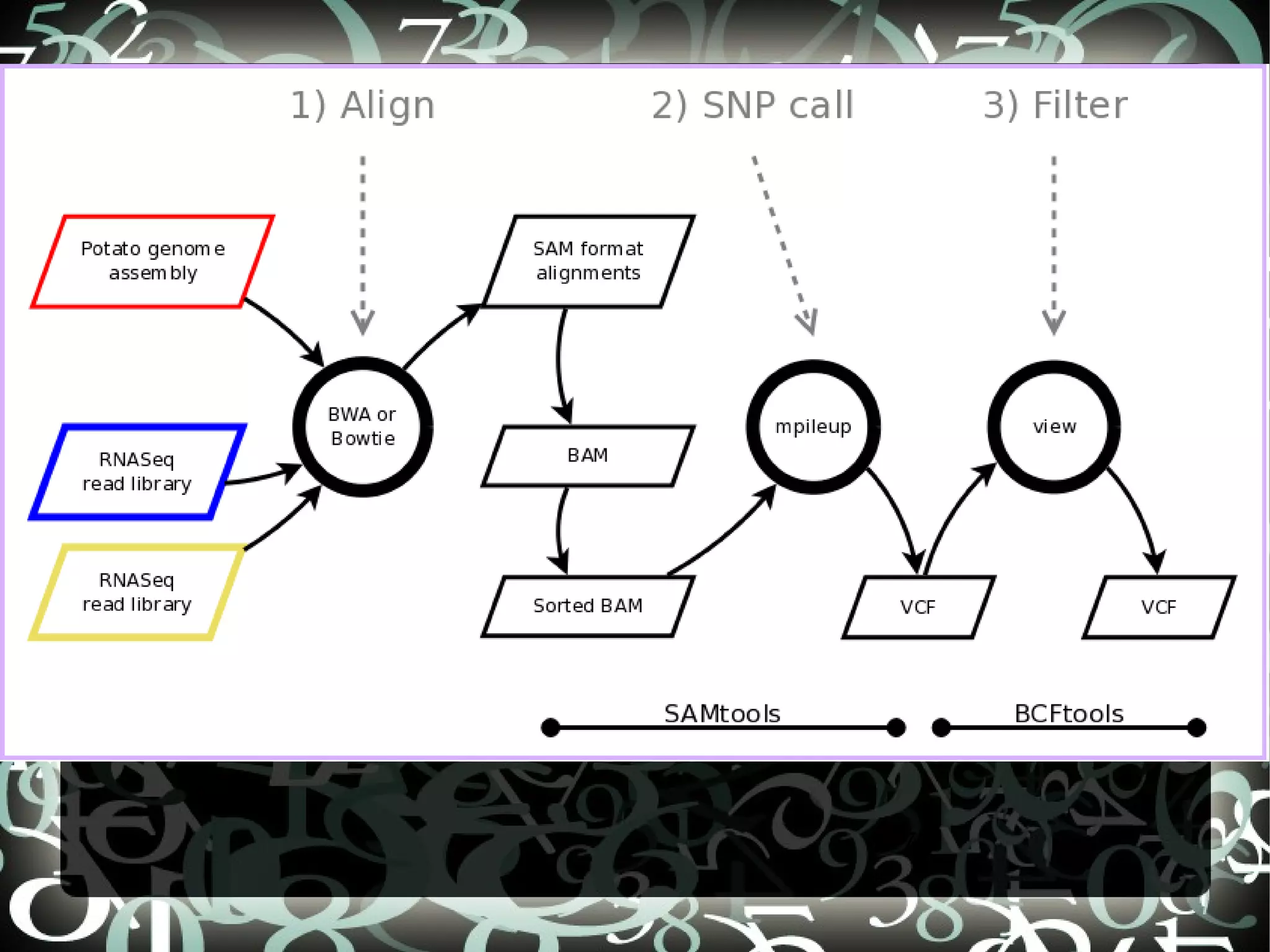

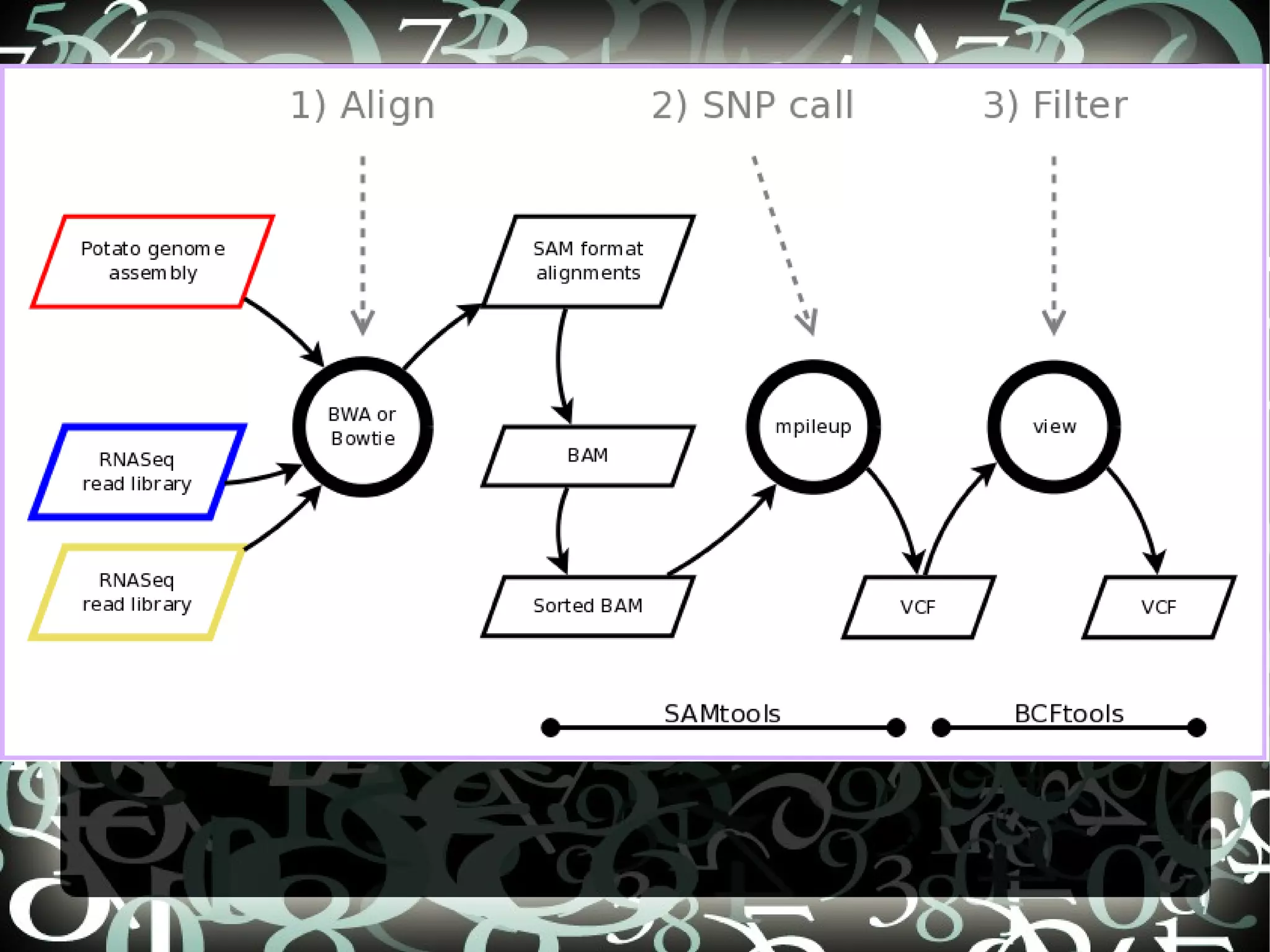



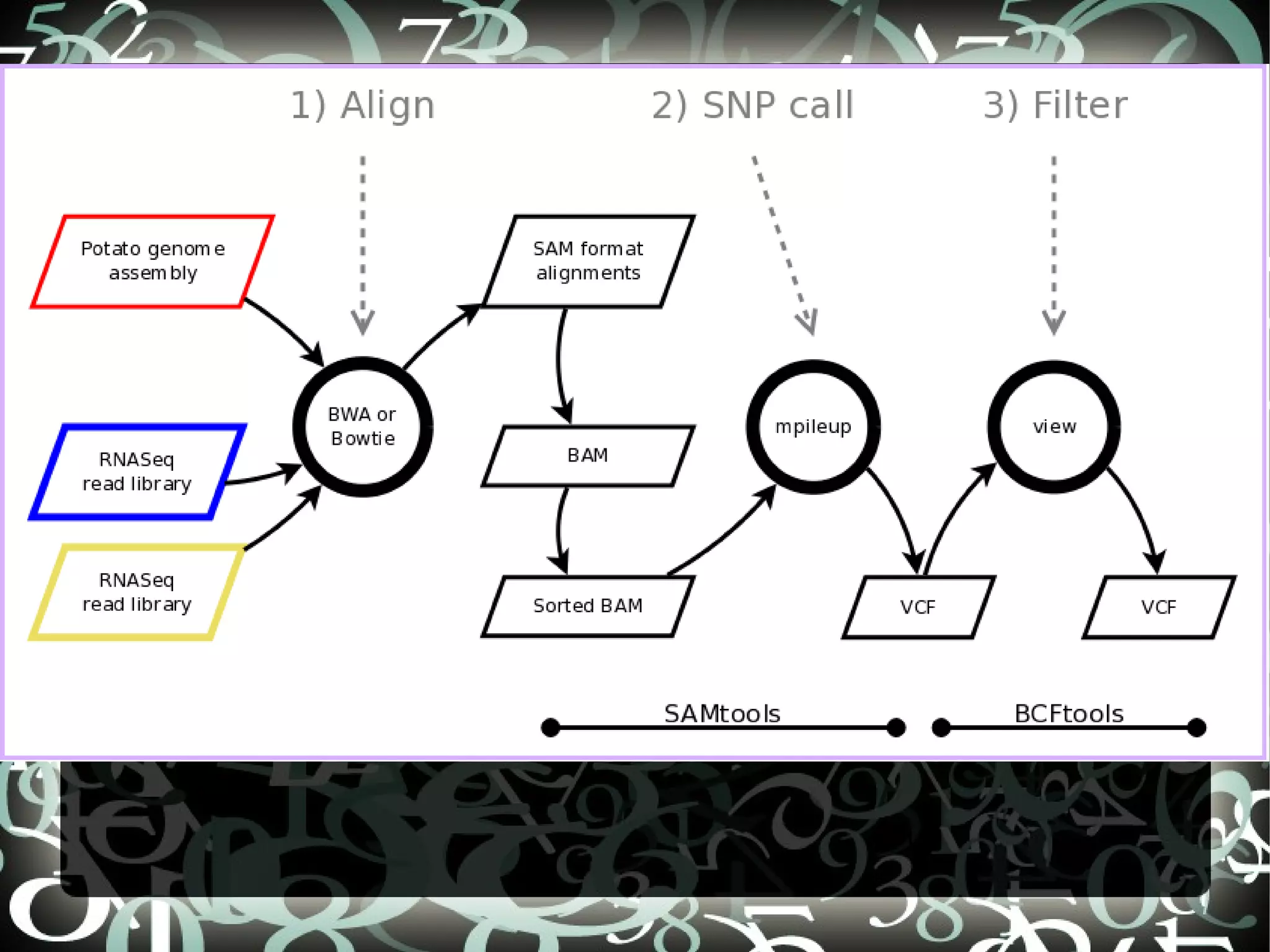




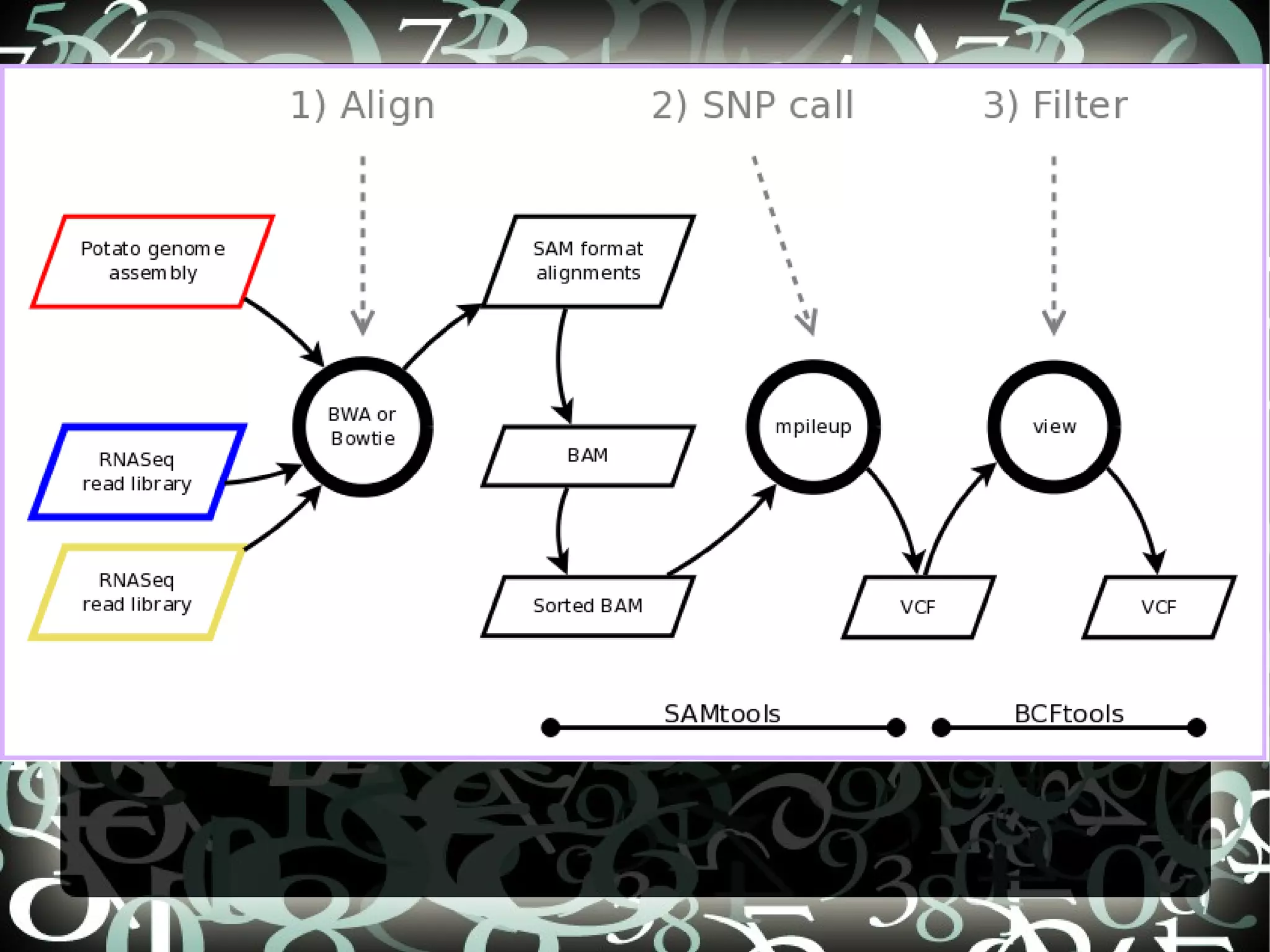
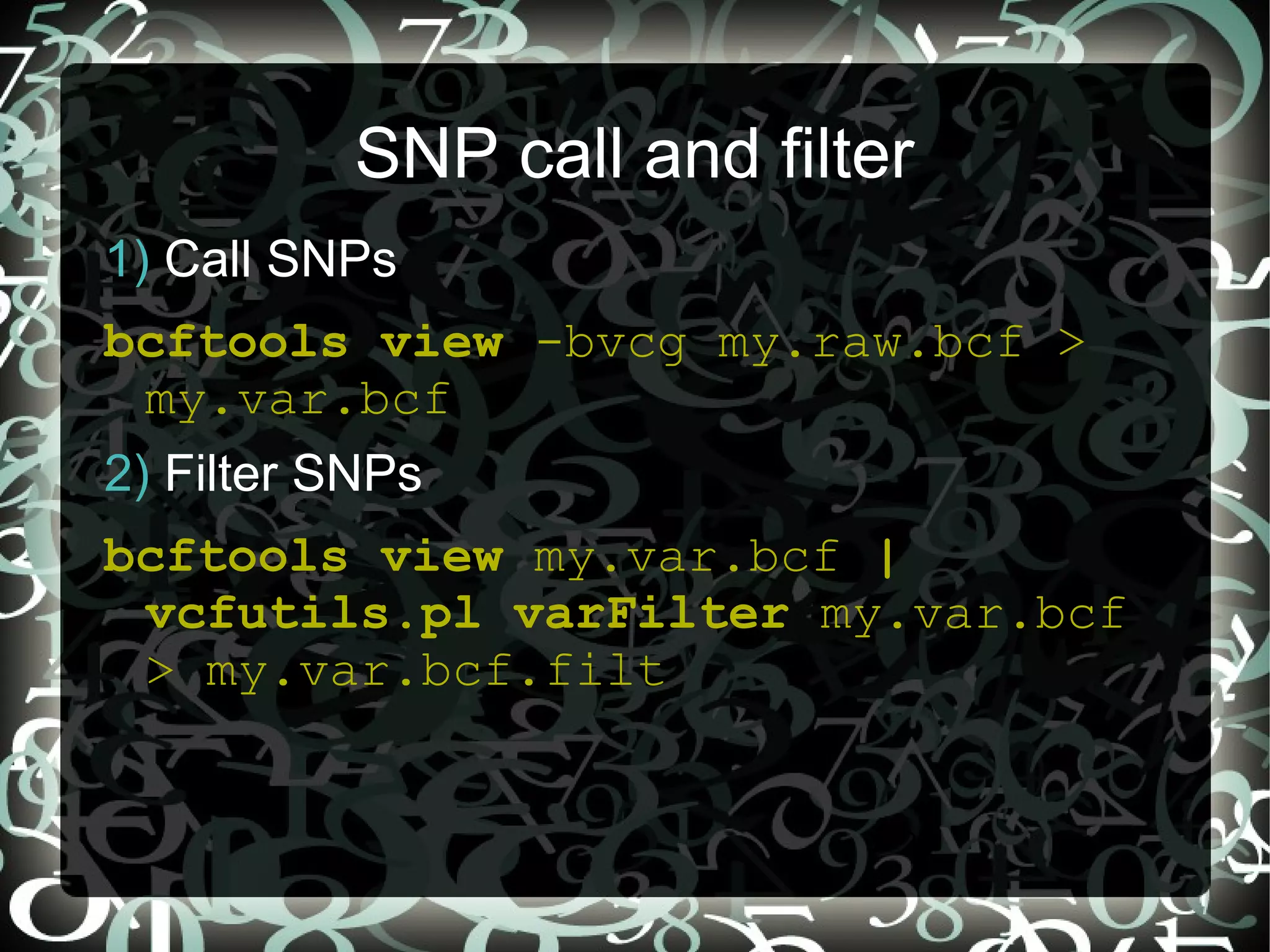
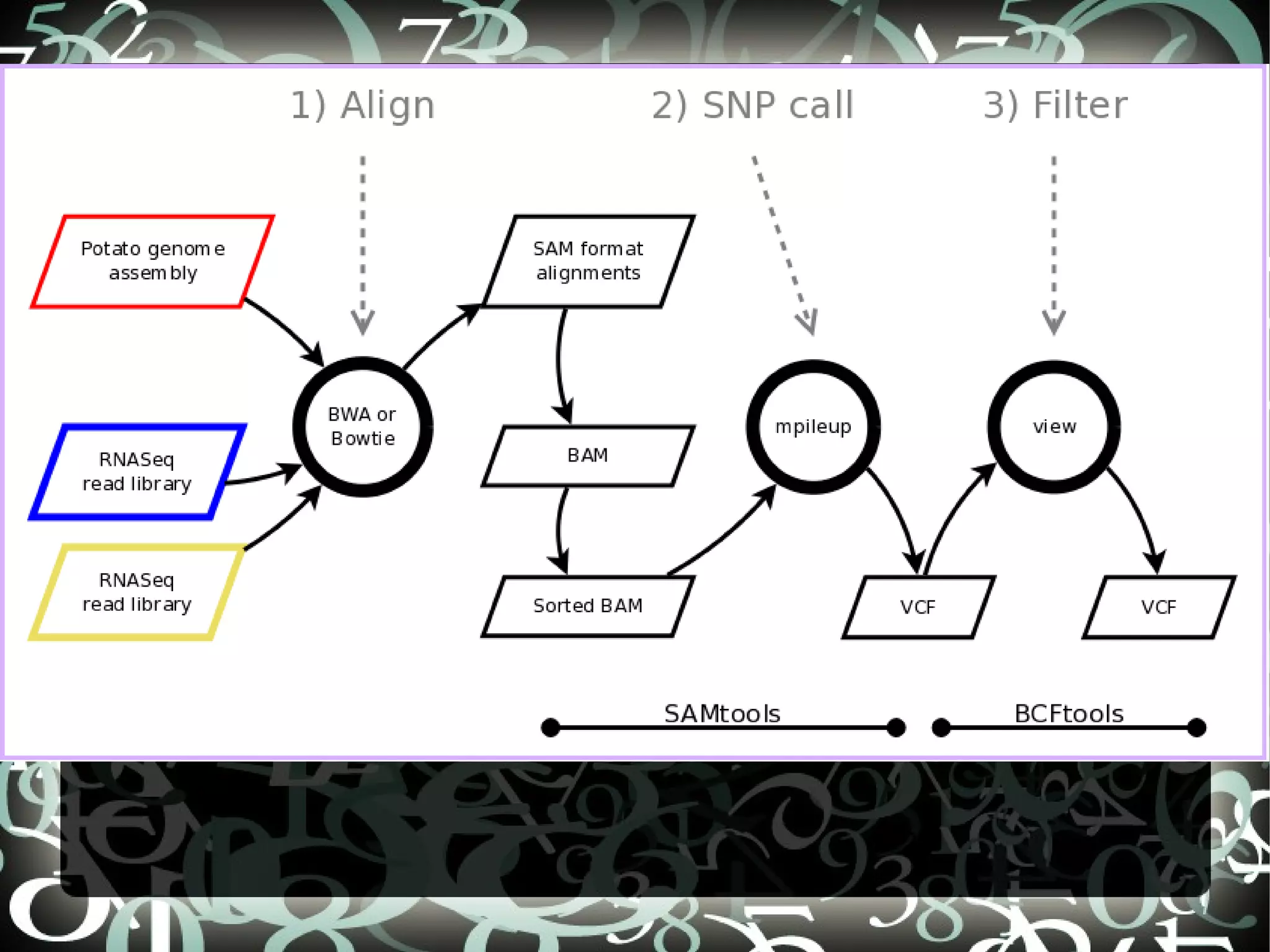





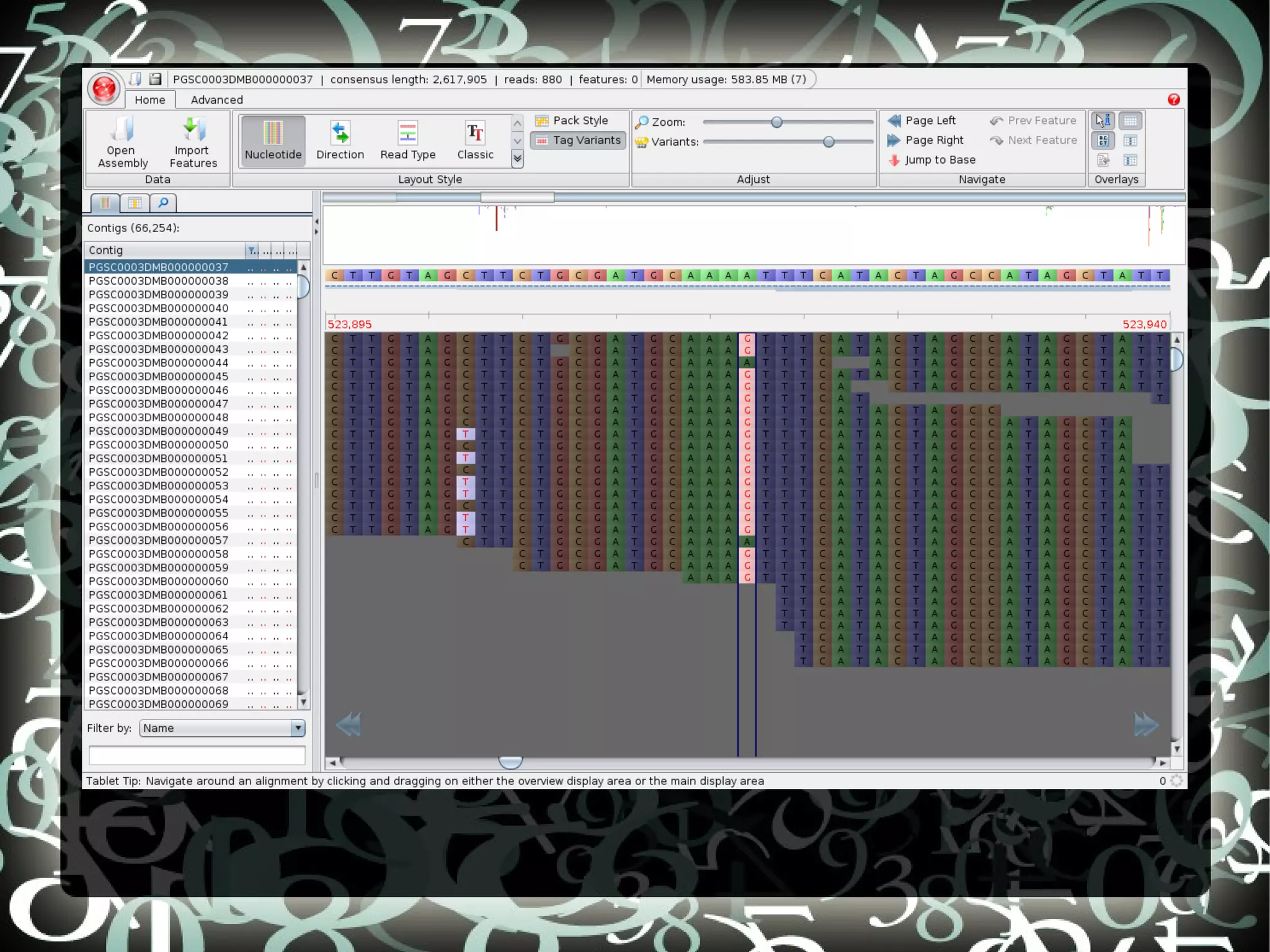
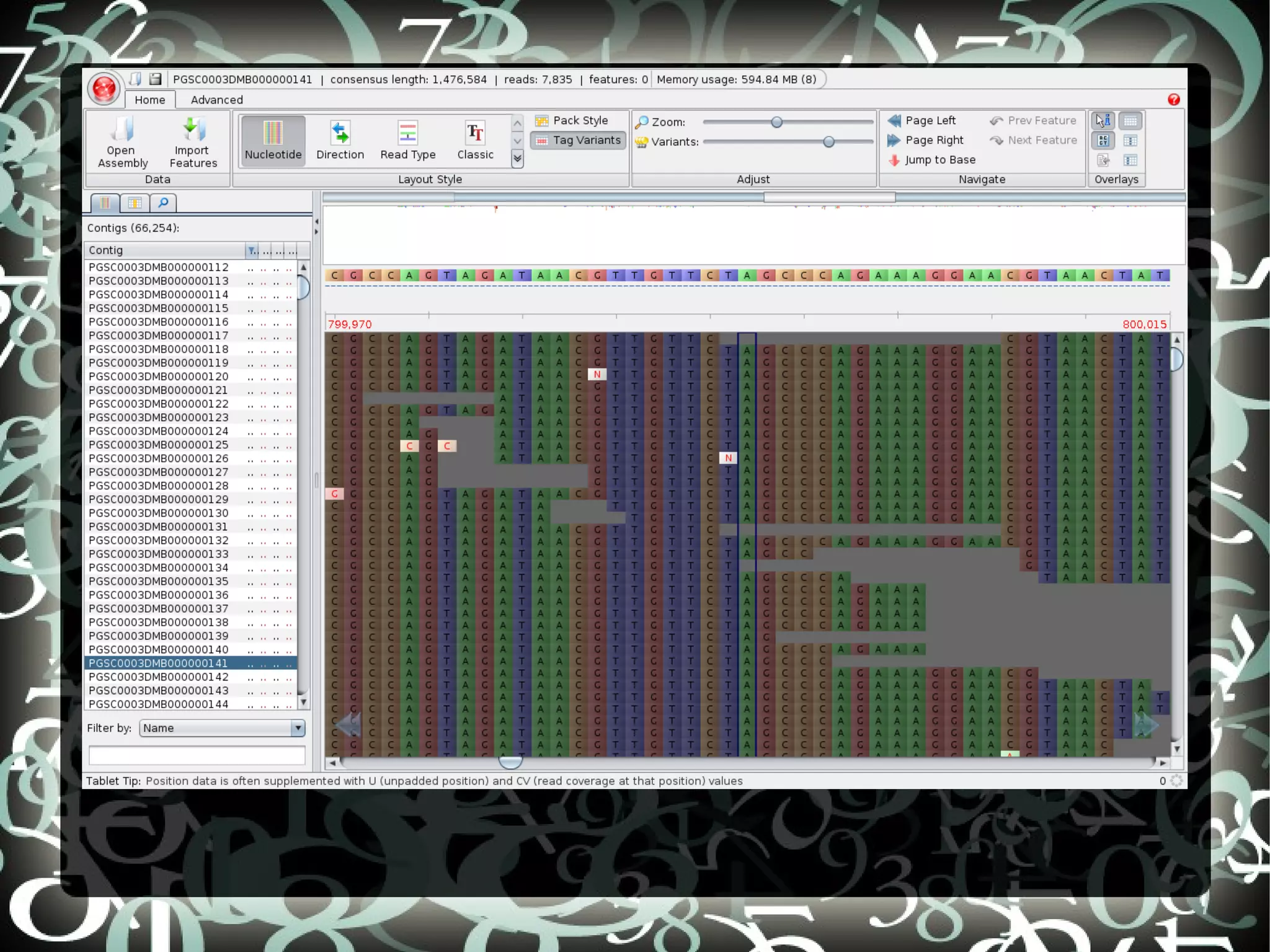


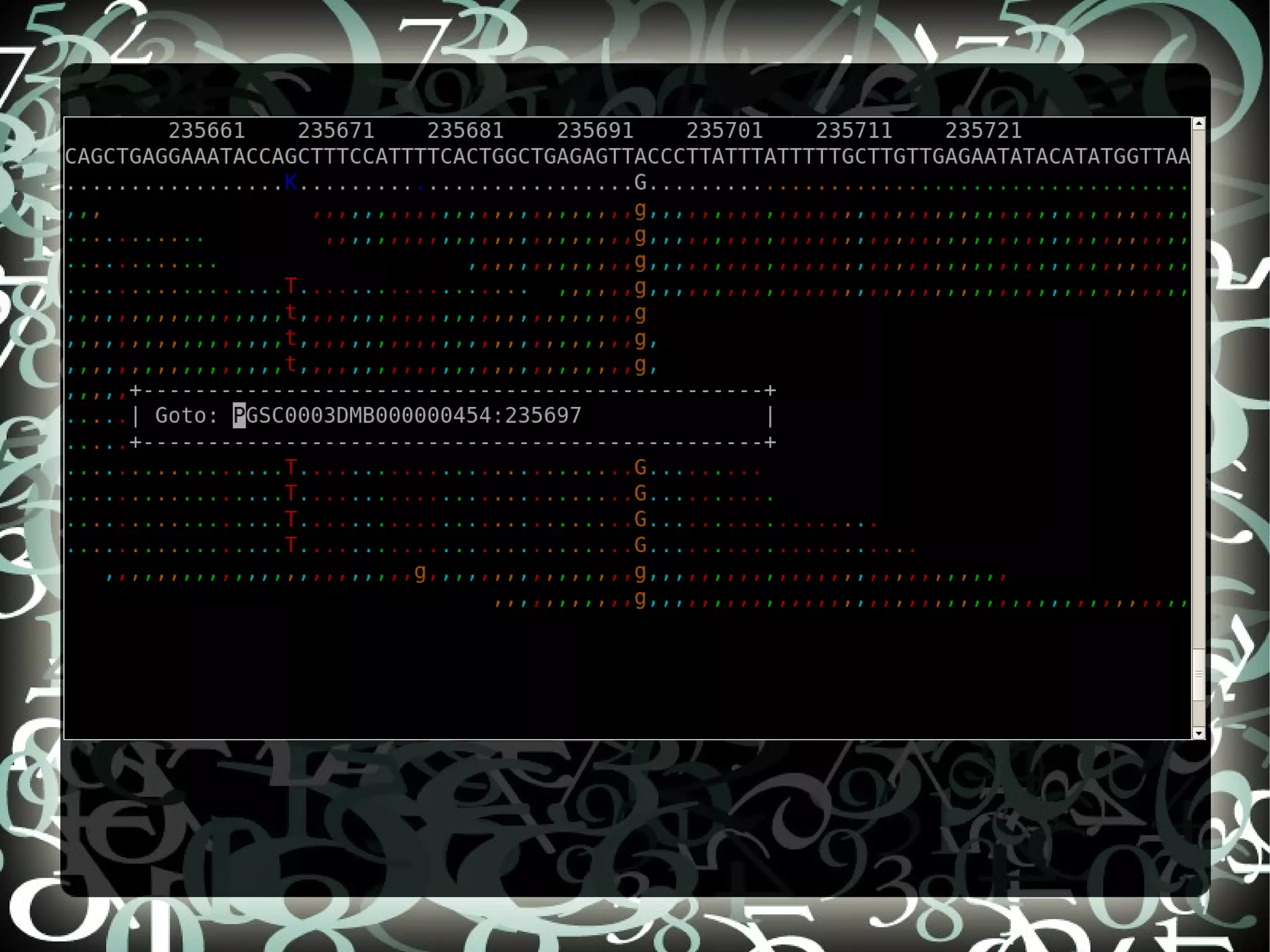
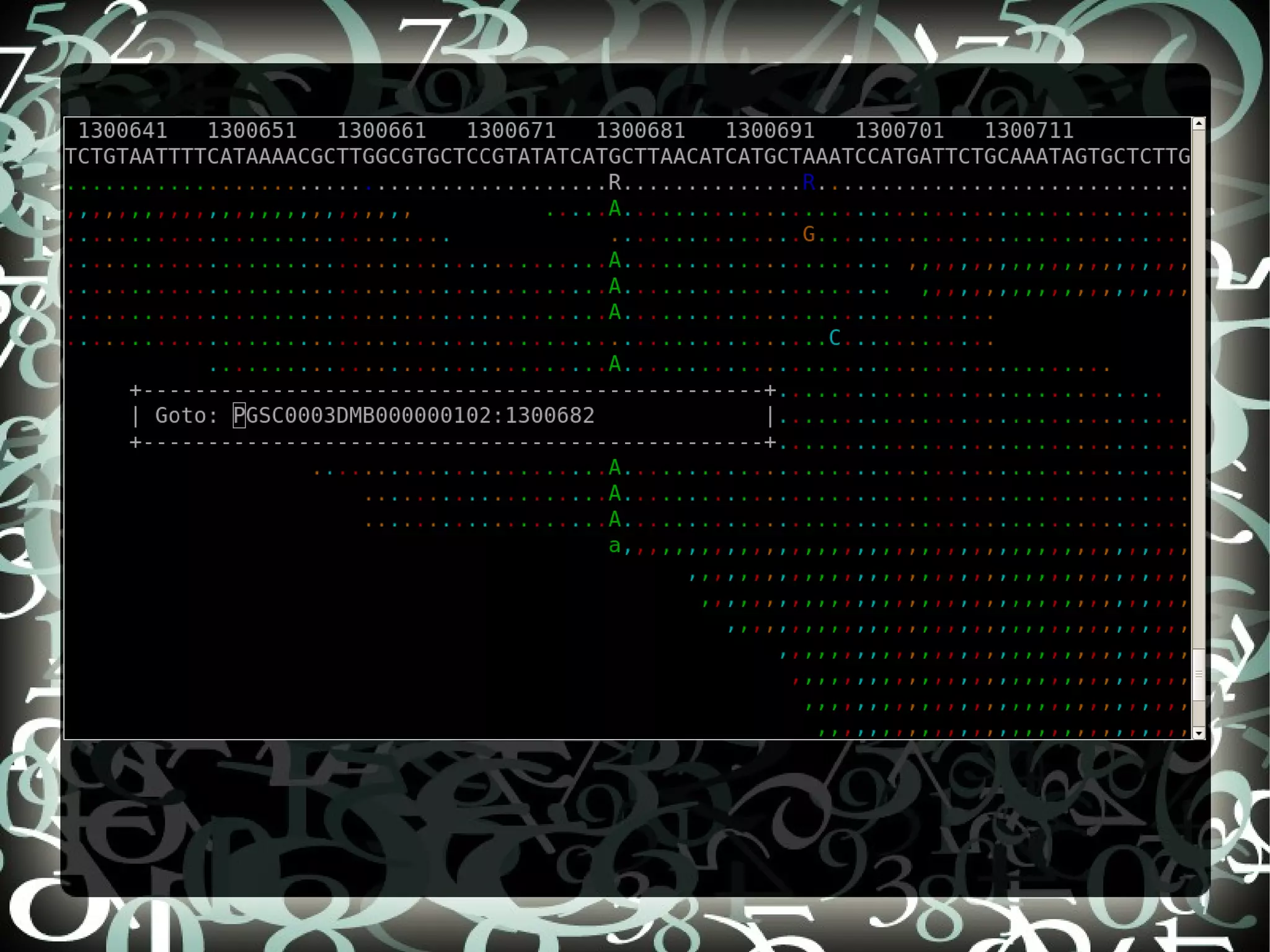


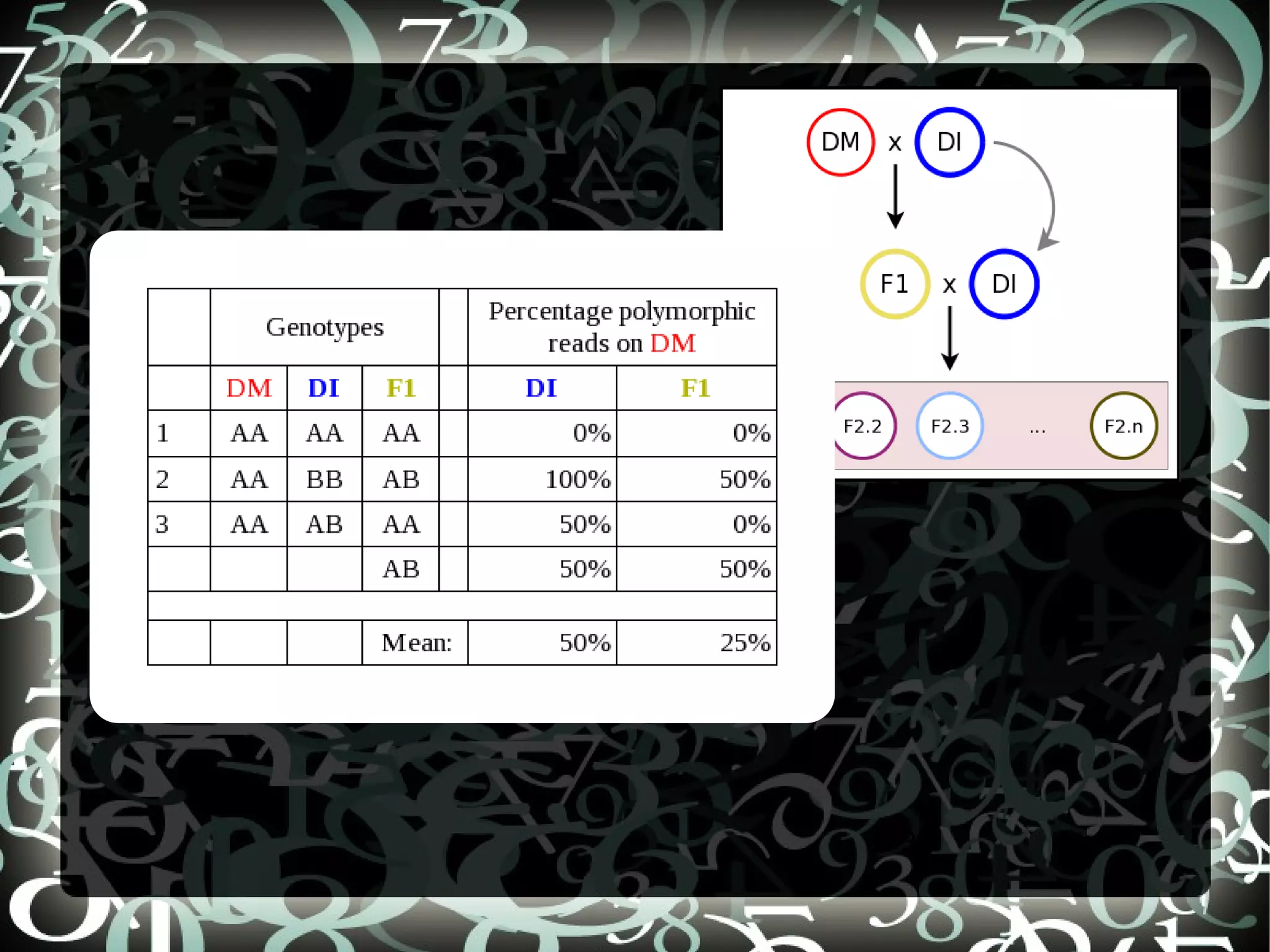
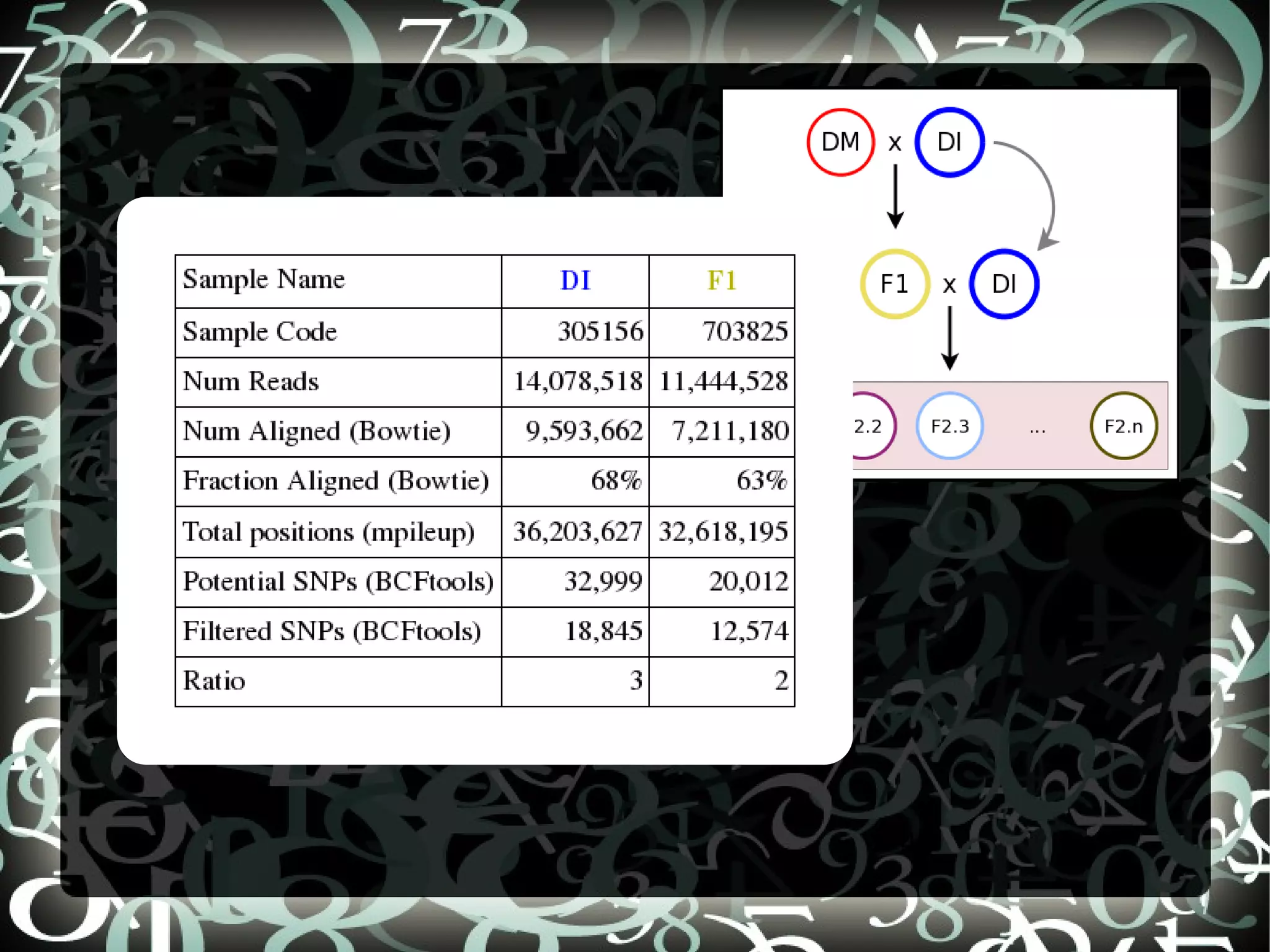
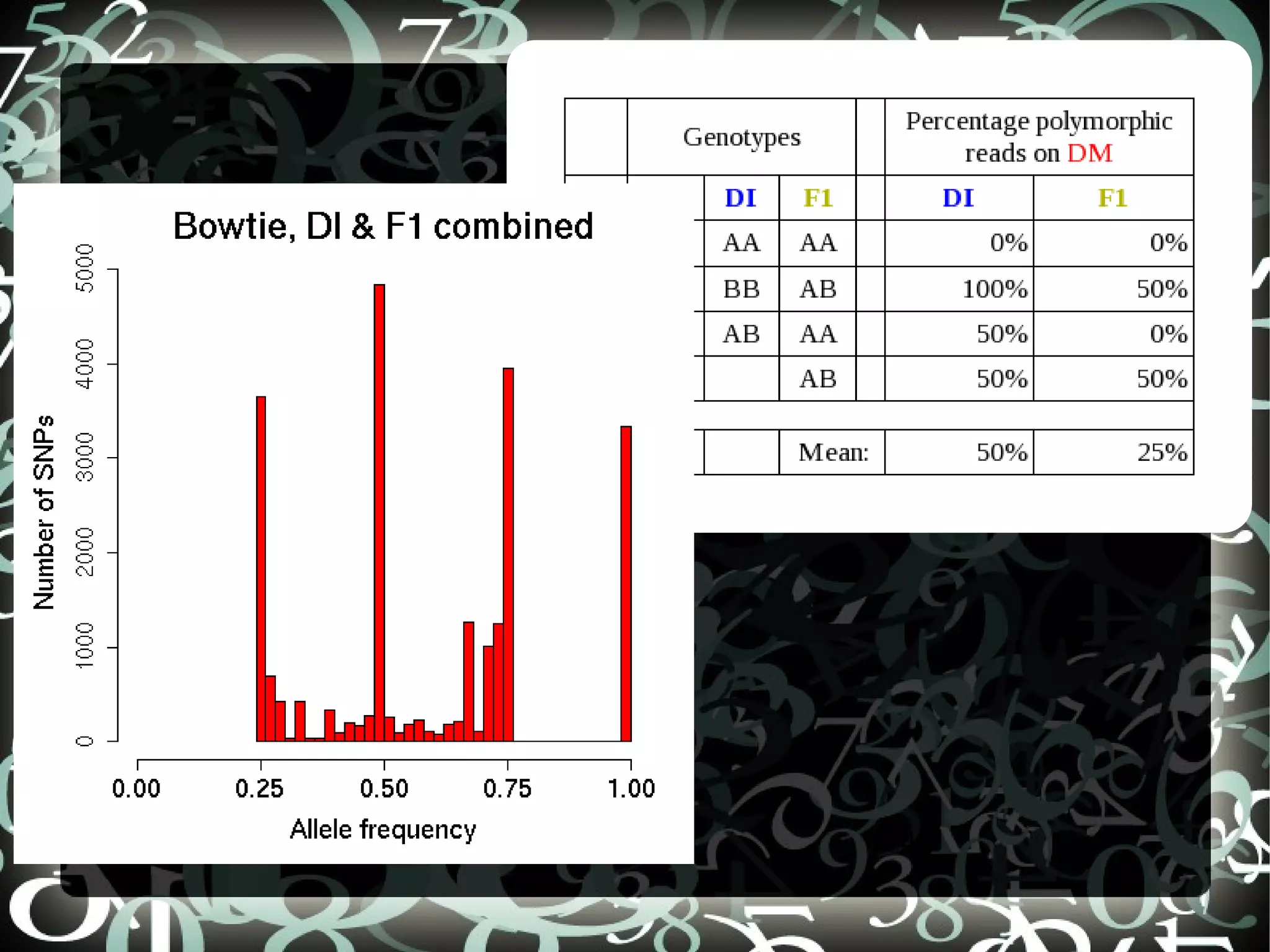







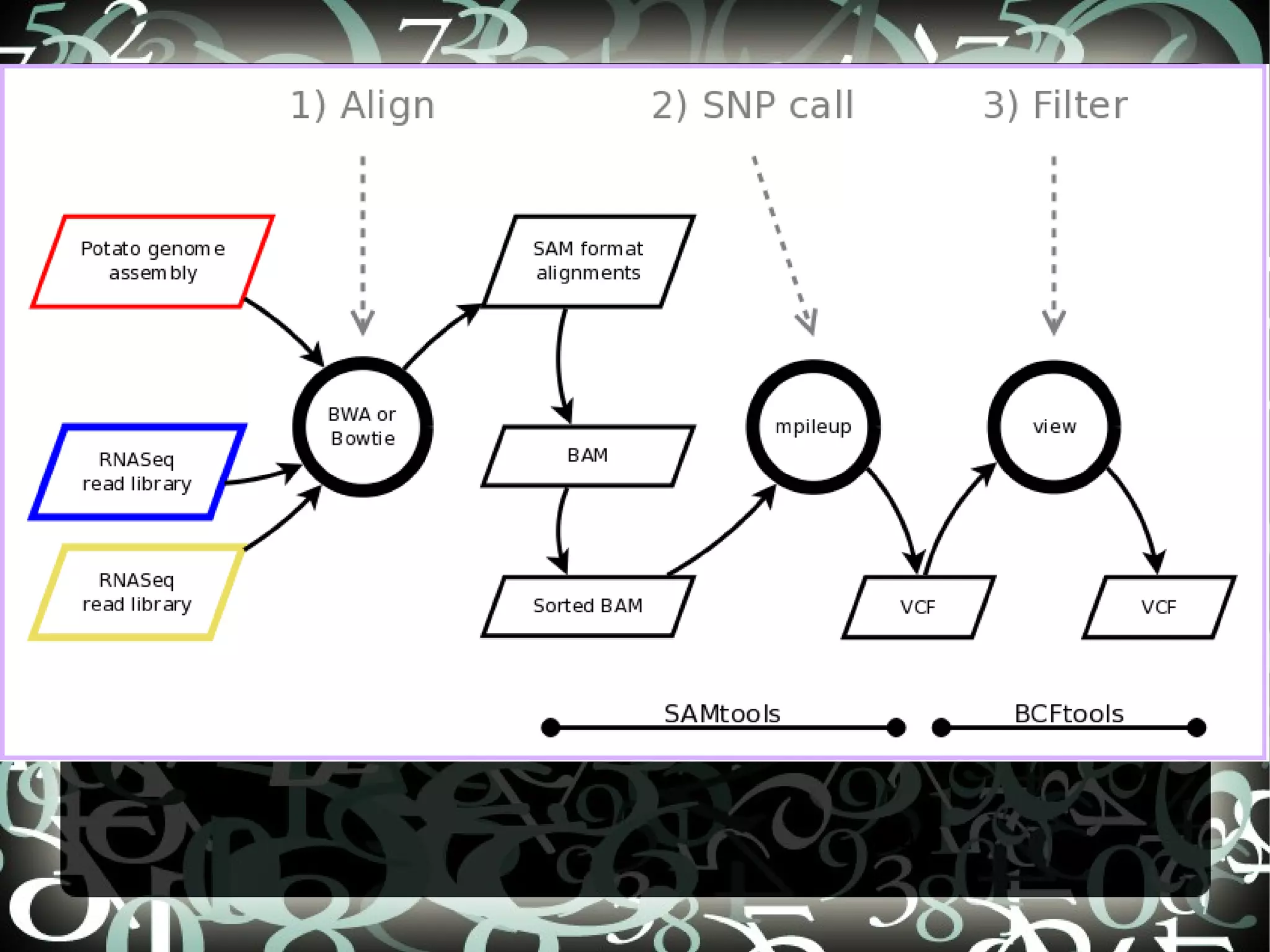
The document describes creating a SNP calling pipeline for potato data from RNA-Seq experiments. Key steps included aligning reads to the potato genome using BWA or Bowtie, converting SAM to BAM and sorting, generating coverage profiles with SAMtools, and calling SNPs from the BAM files using SAMtools and bcftools. SNPs identified from the RNA-Seq data were then selected for inclusion on an Illumina GoldenGate SNP chip to genotype samples for genetic mapping. Comparison of the SNP chip results to the original RNA-Seq data was performed to evaluate accuracy. Remaining questions around discrepancies in the data were noted for further investigation.
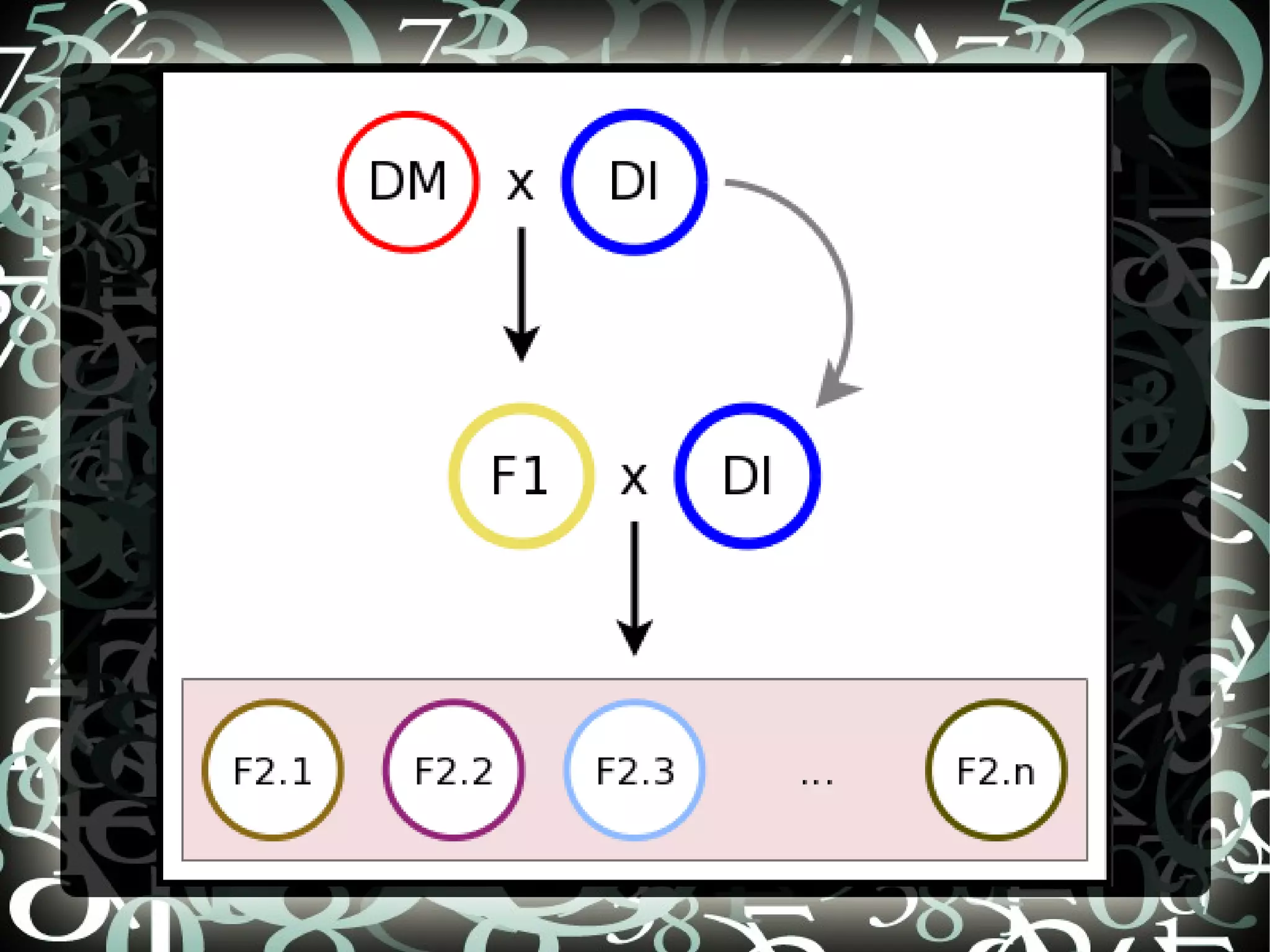
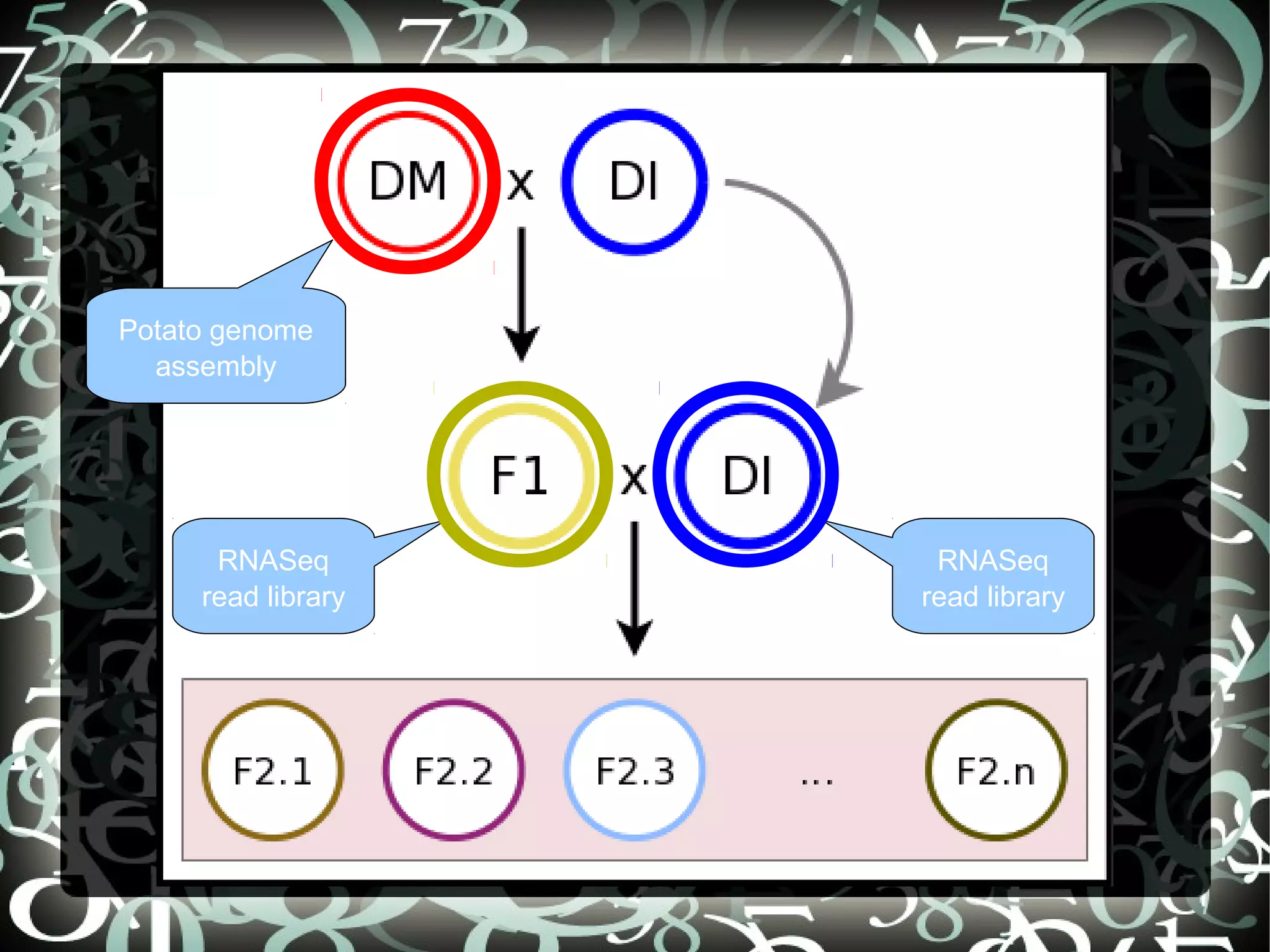
Introduces Potato SNPs and sets aims to create a SNP calling pipeline and select SNPs for genetic mapping using Illumina's GoldenGate SNP chip.
Details steps for creating a SNP calling pipeline, including alignment using BWA and Bowtie, and converting SAM to BAM for SNP calling.
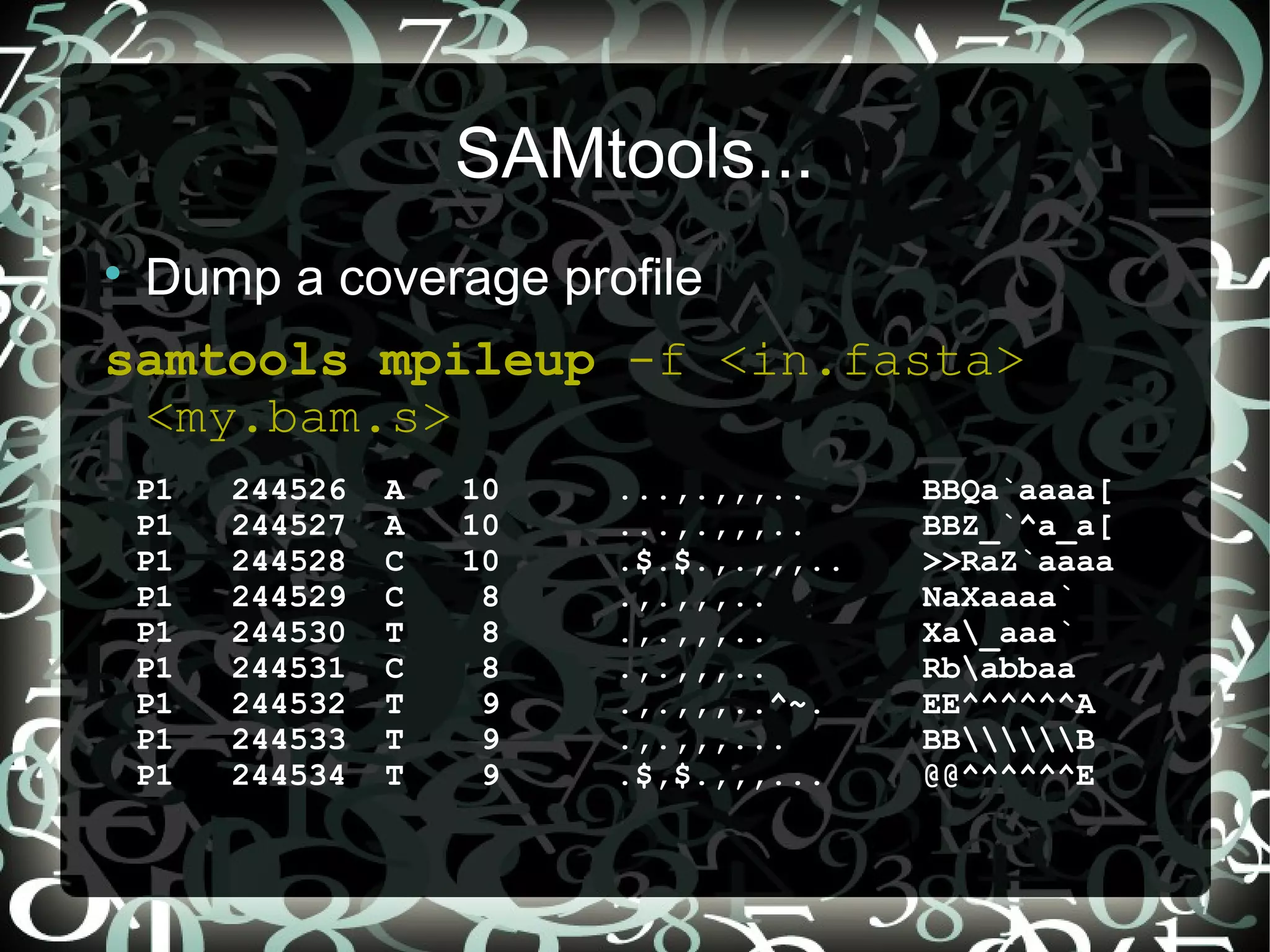
Describes generating coverage profiles using SAMtools, focusing on data presentation and likelihood computation for SNP calling.
Steps to call and filter SNPs using mpileup and BCF format with commands for managing genetic variants.
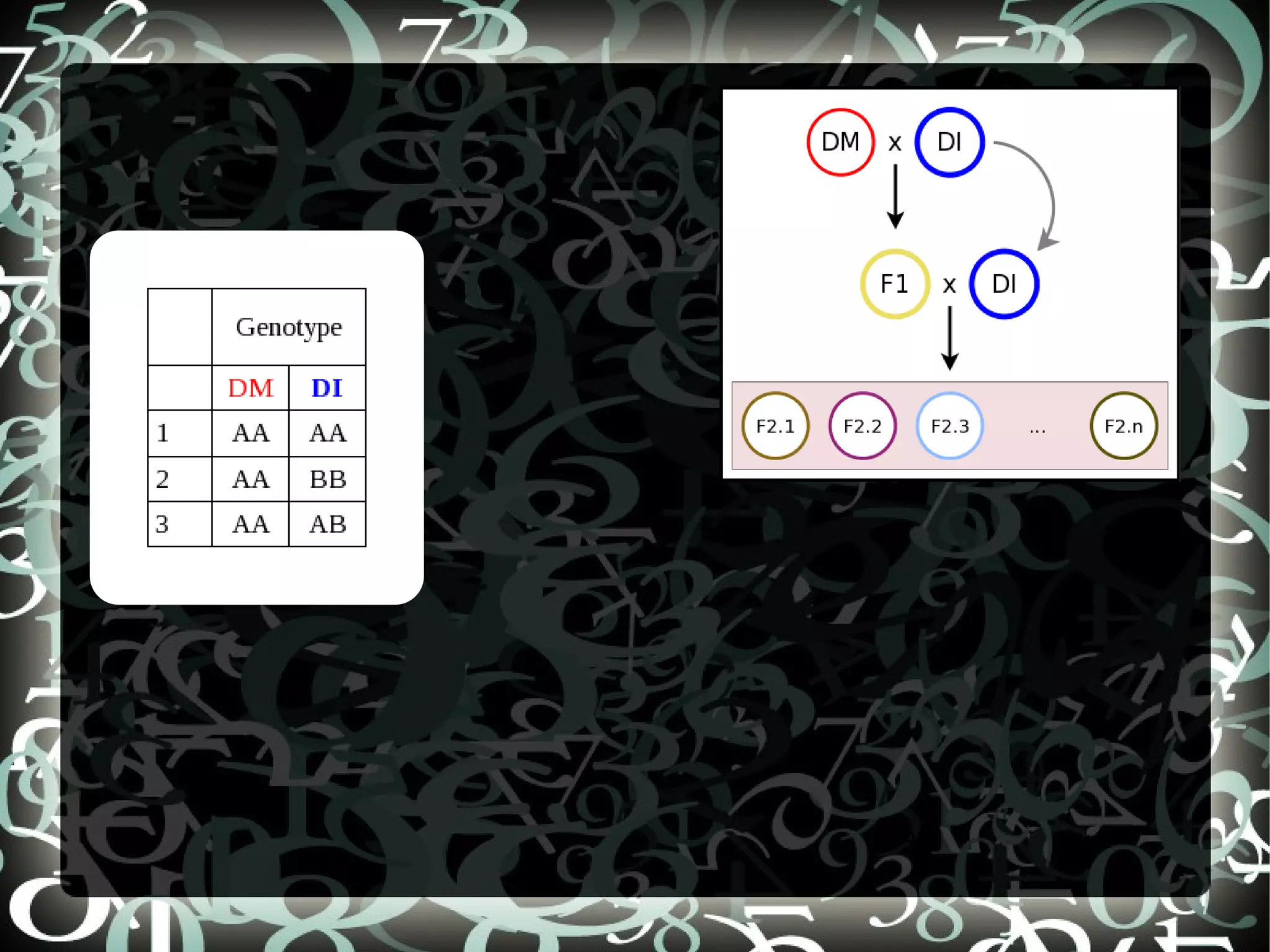
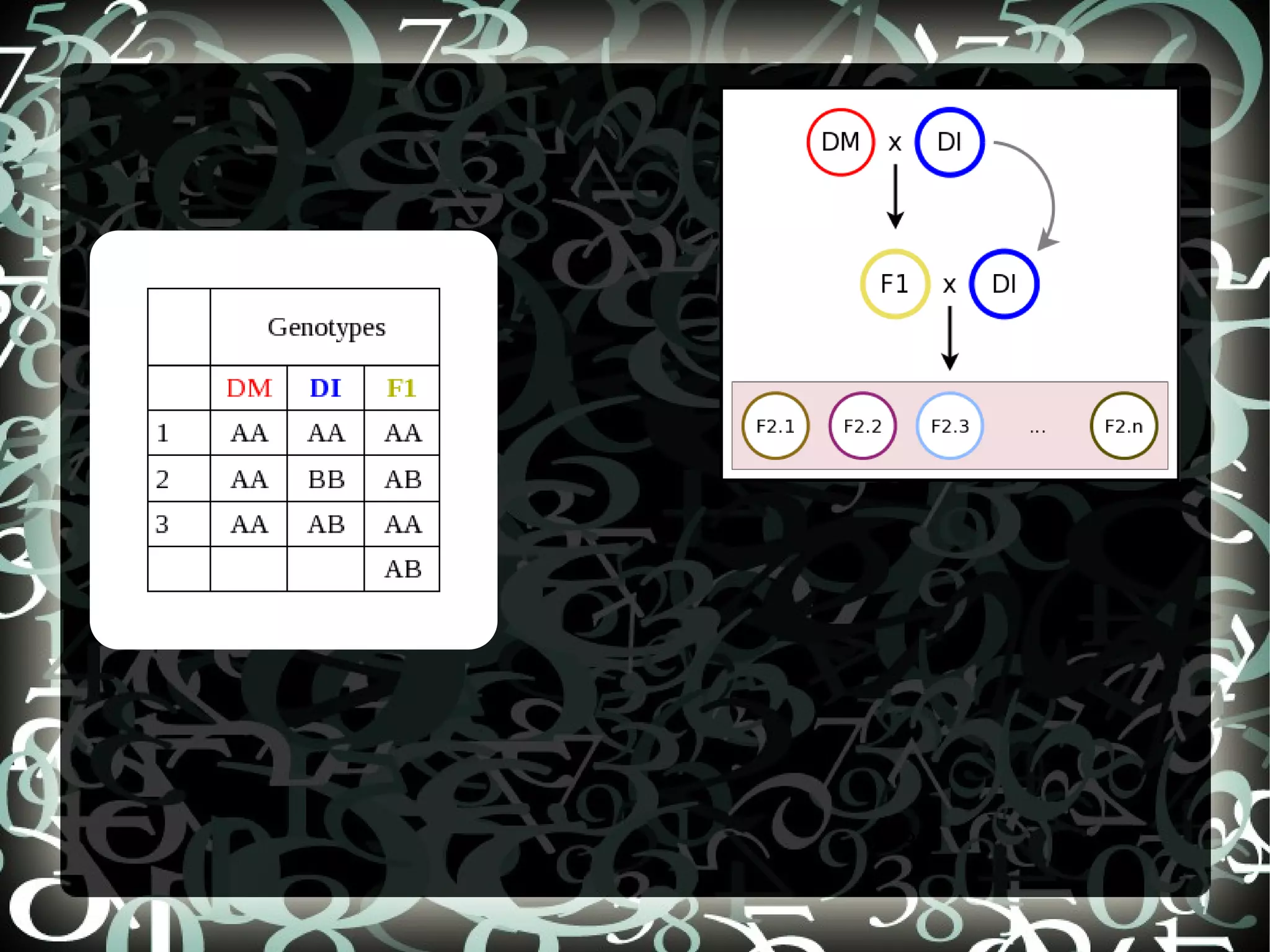
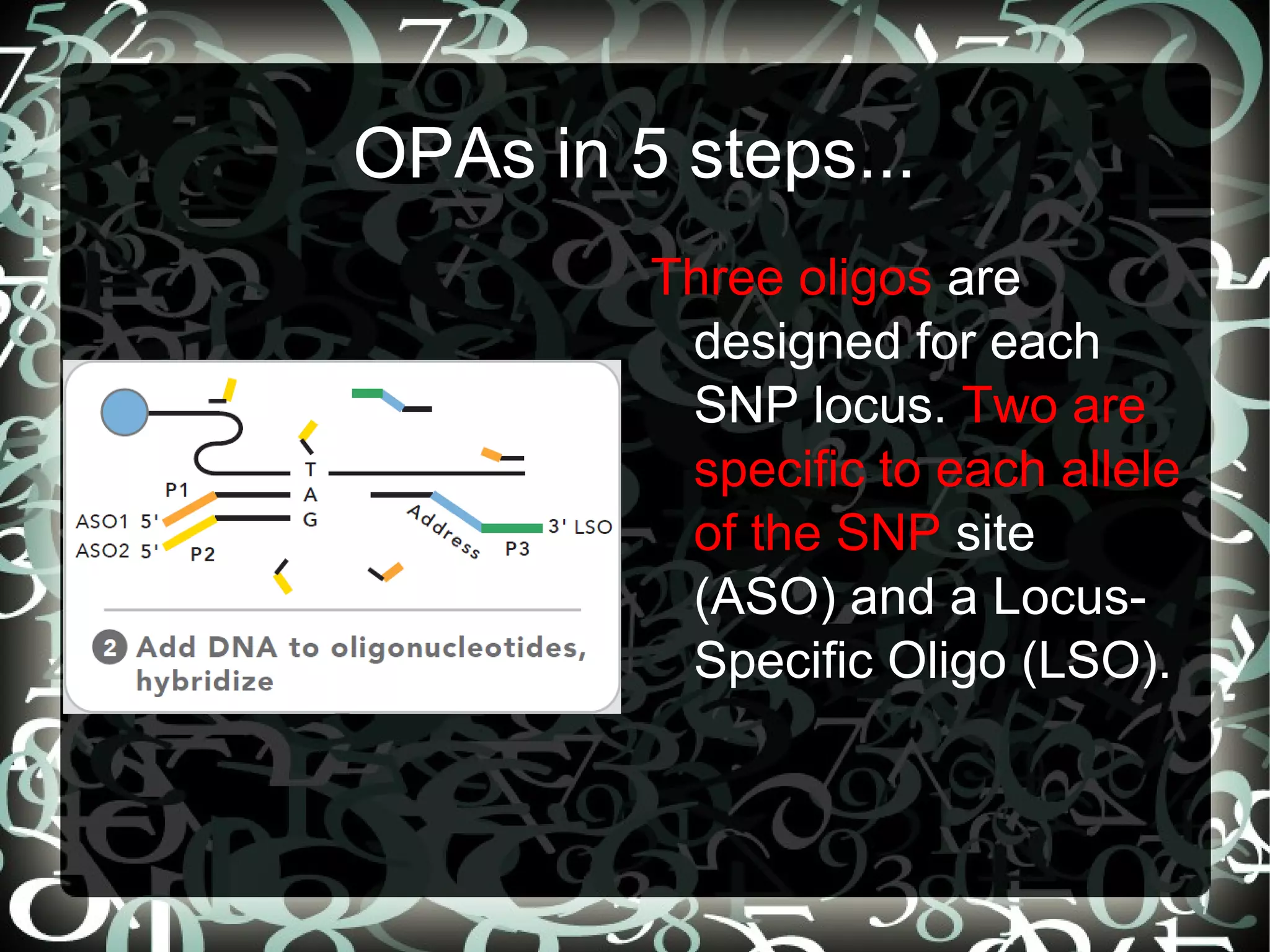
Explains aims of SNP selection and the construction of the SNP chip (OPA) using positions from SolCAP project and comparison to RNASeq.
Compares SNP calls using early and new versions of SAMtools, analyzing similarities in findings through RNASeq data insights.
Discusses questions arising from the study, including exploring atypical SNPs and verifying OPA base correctness.
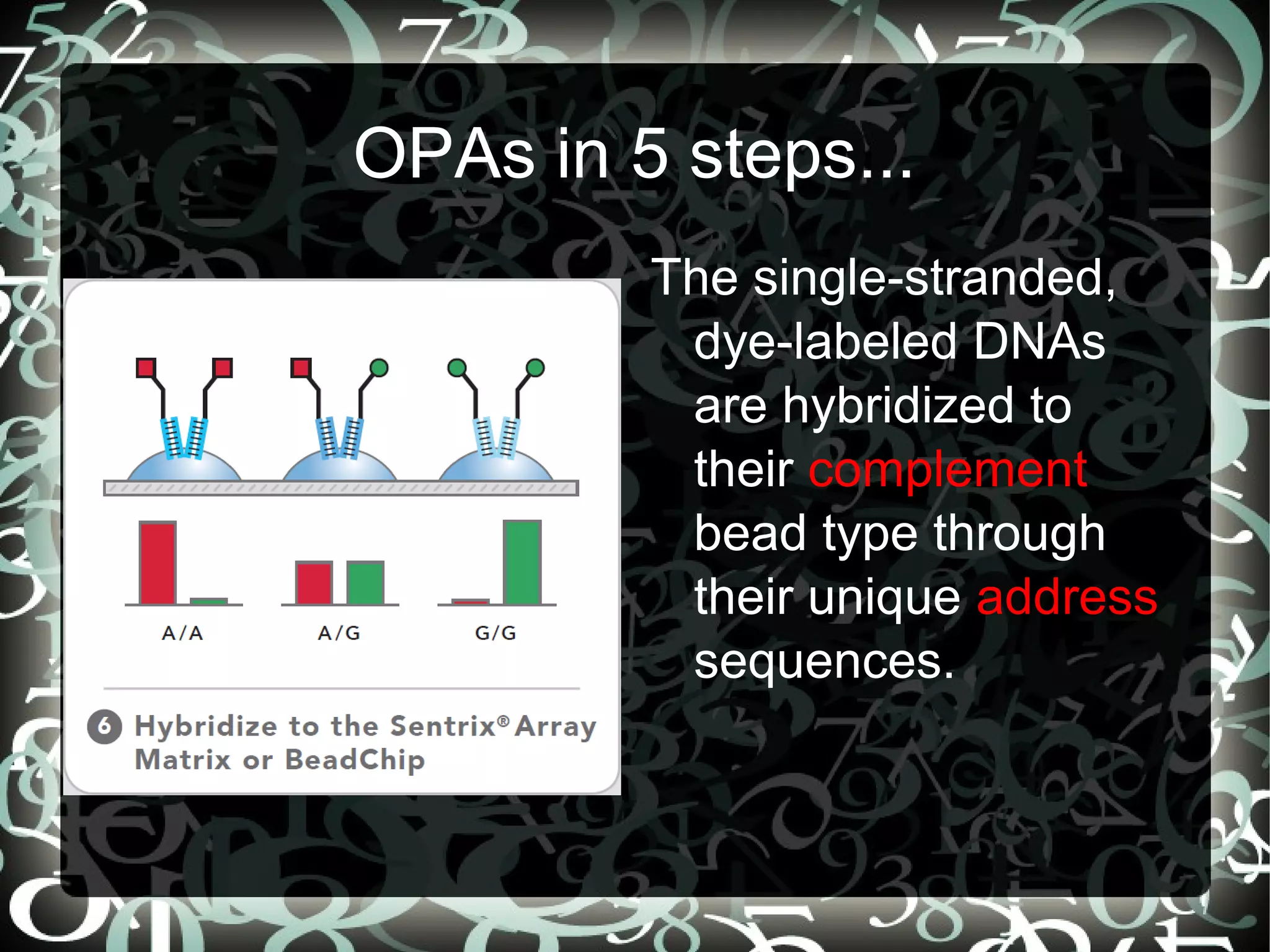
Describes the 5 steps in the Oligonucleotide Pool Assay (OPA) for SNP analysis, highlighting methodology and high accuracy of data.






























































































