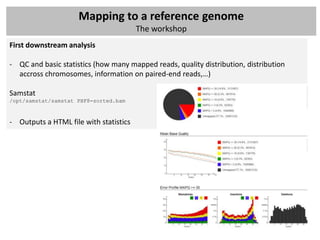
The document outlines a workshop focused on analyzing sequencing data, specifically on mapping raw sequence reads to a reference genome. It details the steps of the mapping process, including the use of various file formats (such as FASTQ, SAM/BAM, and VCF) and specific tools like BWA and SAMtools for alignment and analysis. Additionally, it covers the importance of quality control, statistical analysis, and potential downstream applications such as SNP calling and peak calling.





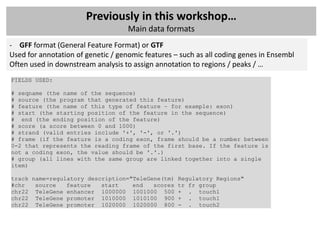
![Previously in this workshop…
Main data formats
- WIG files (location / annotation / scores): wiggle
Used for visulization or summarize data, in most cases count data or normalized count
data (RPKM) – extension: BigWig – binary versions (often used in GEO for ChIP-seq peaks)
browser position chr19:59304200-59310700
browser hide all
#150 base wide bar graph at arbitrarily spaced positions,
#threshold line drawn at y=11.76
#autoScale off viewing range set to [0:25]
#priority = 10 positions this as the first graph
track type=wiggle_0 name="variableStep" description="variableStep format"
visibility=full autoScale=off viewLimits=0.0:25.0 color=50,150,255
yLineMark=11.76 yLineOnOff=on priority=10
variableStep chrom=chr19 span=150
59304701 10.0
59304901 12.5
59305401 15.0
59305601 17.5
59305901 20.0
59306081 17.5](https://image.slidesharecdn.com/20121012sequencing2-121012020608-phpapp02/85/Workshop-NGS-data-analysis-2-9-320.jpg)


![Mapping to a reference genome
The workflow
The reference genome
- Sequences (human; rat: mouse:…) can be downloaded from UCSC (Golden path) or
Ensembl
- Difficulty: download in 2bit format (needs convertor) >> fasta files (.fa)
- Need to be indexed by the mapping program you are going to use
- BWA: bwa index
- Bowtie: bowtie-build (pre-computed indexes available)
- BWA example:
bwa index [-p prefix] [-a algoType] [-c] <in.db.fasta>
Index database sequences in the FASTA format.
OPTIONS:
-c Build color-space index. The input fast should be in nucleotide space.
-p STR Prefix of the output database [same as db filename]
-a STR Algorithm for constructing BWT index. Available options are:
is IS linear-time algorithm for constructing suffix array.
It requires 5.37N memory where N is the size of the database.
bwtsw Algorithm implemented in BWT-SW. This method works with the whole human genome](https://image.slidesharecdn.com/20121012sequencing2-121012020608-phpapp02/85/Workshop-NGS-data-analysis-2-15-320.jpg)

![Mapping to a reference genome
The workflow
aln bwa aln [-n][-o][-e][-d][-i][-k][-l][-t][-cRN][-M][-O][-E][-q]
<in.db.fasta> <in.query.fq> > <out.sai>
Find the SA coordinates of the input reads.
Maximum maxSeedDiff differences are allowed in the first seedLen subsequence
maximum maxDiff differences are allowed in the whole sequence.
OPTIONS:
-n NUM Maximum edit distance if the value is INT
-o INT Maximum number of gap opens
-e INT Maximum number of gap extensions, -1 for k-difference mode
-d INT Disallow a long deletion within INT bp towards the 3’-end
-i INT Disallow an indel within INT bp towards the ends [5]
-l INT Take the first INT subsequence as seed.
-k INT Maximum edit distance in the seed
-t INT Number of threads (multi-threading mode)
-M INT Mismatch penalty
-O INT Gap open penalty
-E INT Gap extension penalty
-R INT Proceed with suboptimal alignments
-c Reverse query but not complement it
-N Disable iterative search.
-q INT Parameter for read trimming.
-I The input is in the Illumina 1.3+ read format (quality equals ASCII-64)
-B INT Length of barcode starting from the 5’-end.
-b Specify the input read sequence file is the BAM format.
-0 When -b is specified, only use single-end reads in mapping.
-1 When -b is specified, only use the first read in a read pair in mapping
-2 When -b is specified, only use the second read in a read pair in mapping](https://image.slidesharecdn.com/20121012sequencing2-121012020608-phpapp02/85/Workshop-NGS-data-analysis-2-17-320.jpg)
![Mapping to a reference genome
The workflow
samse bwa samse [-n maxOcc] <in.db.fasta> <in.sai> <in.fq> > <out.sam>
Generate alignments in the SAM format given single-end reads
Repetitive hits will be randomly chosen.
OPTIONS:
-n INT Maximum number of alignments to output in the XA tag for reads paired properly.
-r STR Specify the read group in a format like ‘@RGtID:footSM:bar’
sampe bwa sampe [-a][-o][-n][-N][-P]<in.db.fasta>
<in1.sai><in2.sai><in1.fq><in2.fq> ><out.sam>
Generate alignments in the SAM format given paired-end reads.
Repetitive read pairs will be placed randomly.
OPTIONS:
-a INT Maximum insert size for a read pair to be considered being mapped properly.
-o INT Maximum occurrences of a read for pairing.
-P Load the entire FM-index into memory to reduce disk operations
-n INT Maximum number of alignments to output in the XA tag for reads paired properly
-N INT Maximum number of alignments to output in the XA tag for disconcordant read pairs
-r STR Specify the read group in a format like ‘@RGtID:footSM:bar’](https://image.slidesharecdn.com/20121012sequencing2-121012020608-phpapp02/85/Workshop-NGS-data-analysis-2-18-320.jpg)