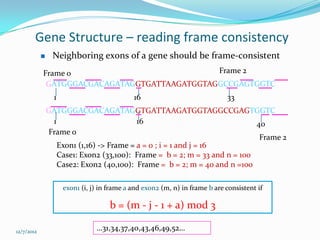
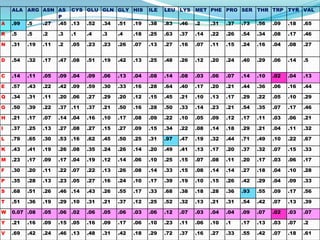
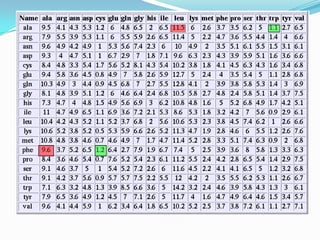
The document discusses topics to be covered in an IICB course on 8th December 2012, including primer designing, restriction mapping, and gene prediction. It provides information and guidelines on these topics, such as the appropriate length and properties of primers, the four types of restriction enzymes, and methods for gene prediction including patterns, frame consistency, dicodon frequencies, position-specific scoring matrices, and coding potential. Relevant references and websites discussing these techniques are also listed.