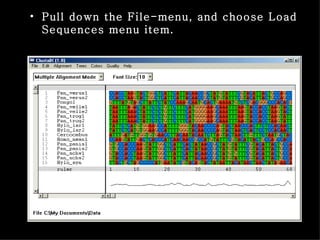
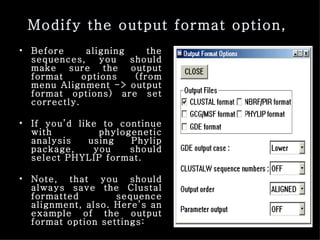
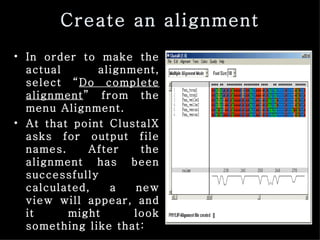
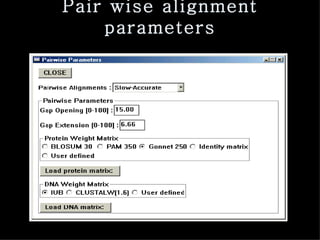
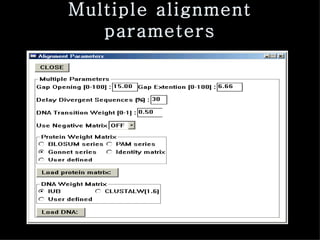
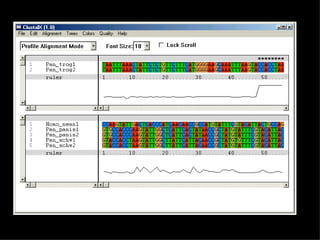
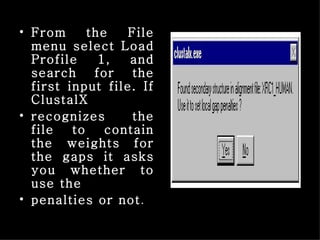
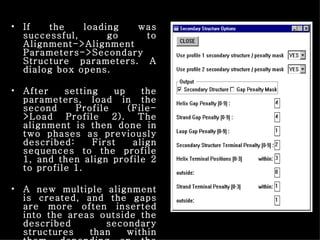
ClustalX is a Windows interface for the ClustalW multiple sequence alignment program, which enables users to perform and analyze multiple sequence alignments. It supports various input formats and provides options for profile alignment, incorporating secondary structure information to adjust gap penalties. The program uses a progressive method, relying on pairwise alignments and phylogenetic trees to achieve global multiple sequence alignments.