Downloaded 58 times





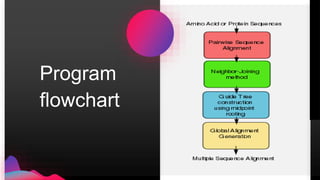

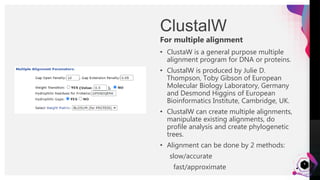
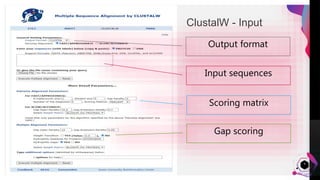

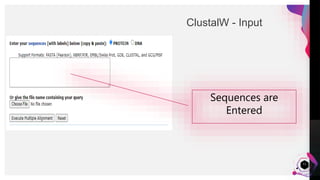

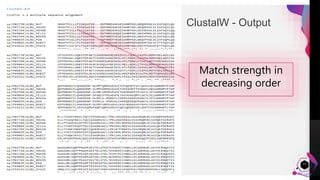
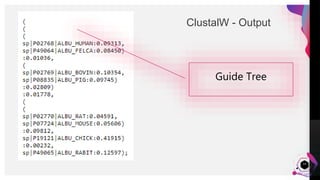
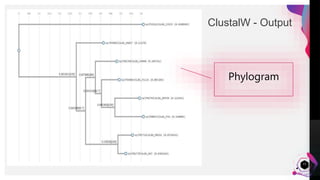




The document provides an overview of ClustalW, a bioinformatics tool used for aligning multiple nucleotide or protein sequences through progressive alignment methods. It includes an introduction to KEGG and Genomenet, as well as details on the algorithm and workflow of ClustalW for multiple sequence alignment. Additionally, it mentions related tools such as Clustalω and Jalview, and provides resources for further information.



































































