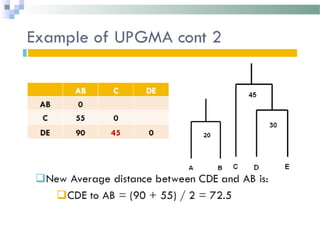
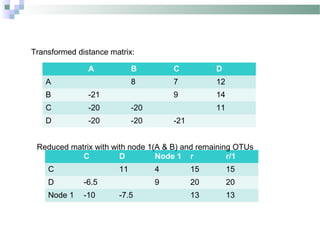
Phylogenetic analyses involve constructing evolutionary trees to model the relationships between taxa. There are two main components: 1) tree building to infer branching orders and evolutionary relationships between taxa, and 2) character and rate analysis using trees as frameworks to understand trait evolution. Trees are constructed using clustering algorithms and optimality criteria applied to distance or character data. Parsimony methods infer the most parsimonious tree requiring the fewest evolutionary events, and can be used to analyze molecular and non-molecular data as well as ancestral sequence inference.











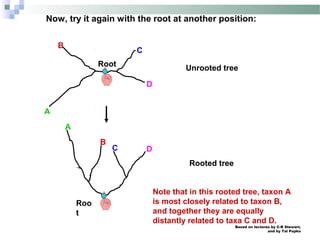



![Based on lectures by C-B Stewart,
and by Tal Pupko
Parsimony methods:
Optimality criterion: The ‘most-parsimonious’ tree is the one that
requires the fewest number of evolutionary events (e.g., nucleotide
substitutions, amino acid replacements) to explain the sequences.
Advantages:
• Are simple, intuitive, and logical (many possible by ‘pencil-and-paper’).
• Can be used on molecular and non-molecular (e.g., morphological) data.
• Can tease apart types of similarity (shared-derived, shared-ancestral, homoplasy)
• Can be used for character (can infer the exact substitutions) and rate analysis.
• Can be used to infer the sequences of the extinct (hypothetical) ancestors.
Disadvantages:
• Are simple, intuitive, and logical (derived from “Medieval logic”, not statistics!)
• Can be fooled by high levels of homoplasy (‘same’ events).
• Can become positively misleading in the “Felsenstein Zone”:
[See Stewart (1993) for a simple explanation of parsimony analysis, and Swofford
et al. (1996) for a detailed explanation of various parsimony methods.]](https://image.slidesharecdn.com/phylogeneticanalyses1-180504172001/85/Phylogenetic-analyses1-16-320.jpg)