Downloaded 440 times





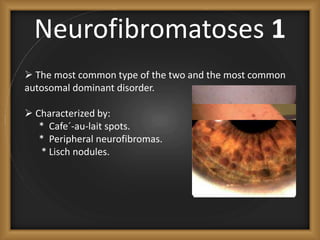
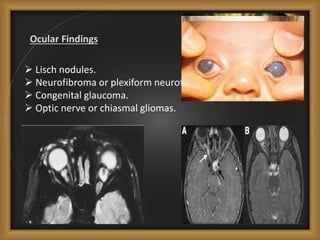
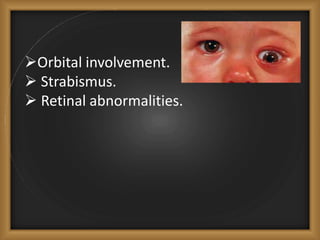
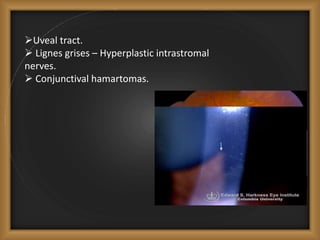
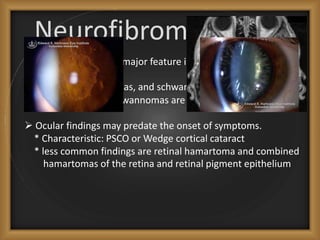

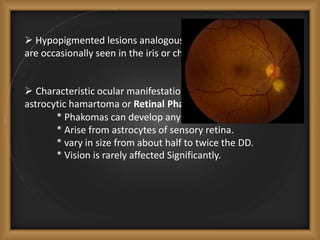

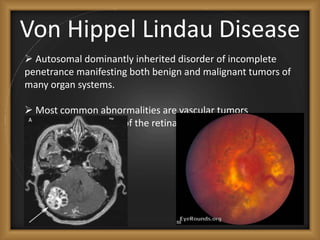

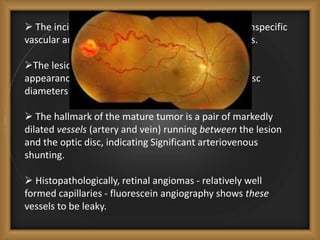

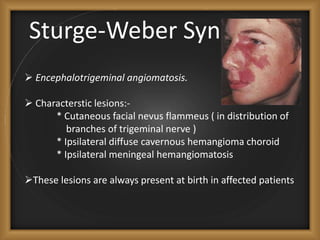
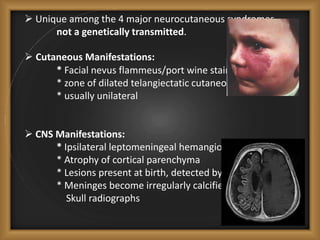
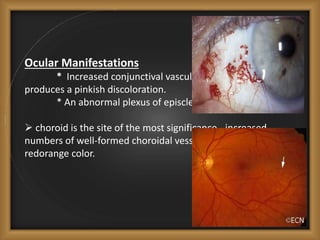



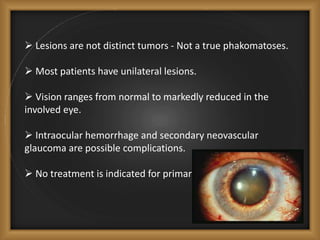
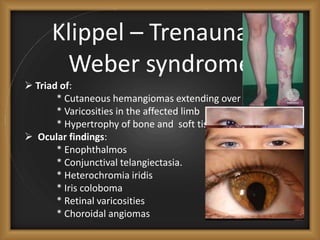
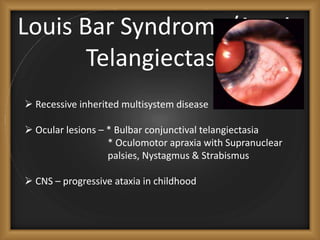
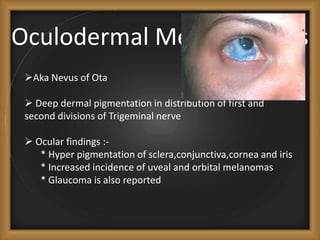
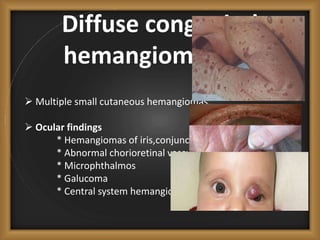
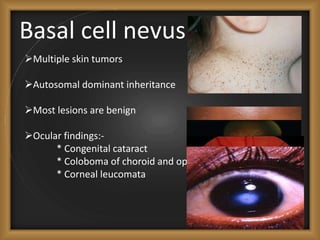

Phacomatoses are syndromes characterized by hamartomas affecting the skin, eyes, CNS, and other organs, leading to significant visual and neurological disturbances, often due to single-gene mutations. Common conditions include neurofibromatosis types 1 and 2, tuberous sclerosis, Sturge-Weber syndrome, and von Hippel-Lindau disease, each presenting distinct ocular manifestations and complications. Management varies by syndrome, with early diagnosis and treatment crucial for favorable outcomes, particularly in conditions like von Hippel-Lindau disease, where retinal lesions can lead to vision loss.














































![Myasthenia gravis guest_lecture[1]](https://cdn.slidesharecdn.com/ss_thumbnails/myastheniagravisguestlecture1-120720130546-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)












































![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)









