Downloaded 894 times


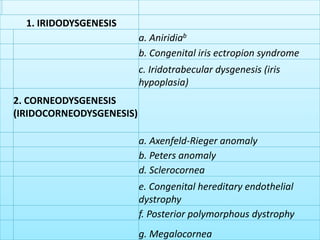


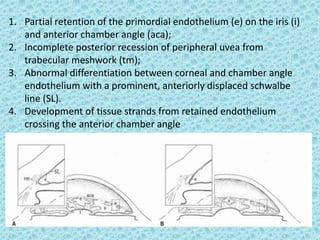

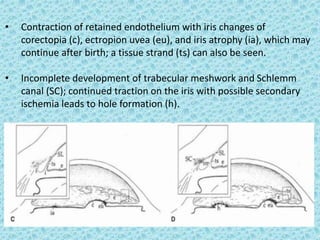

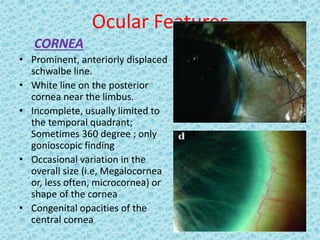


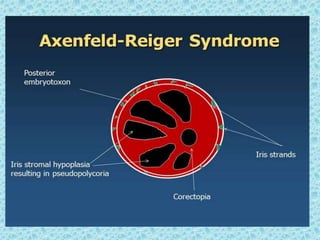


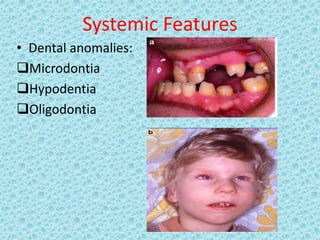

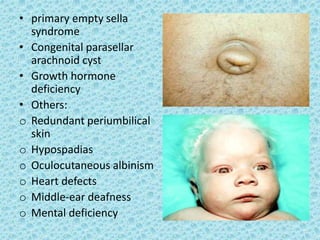



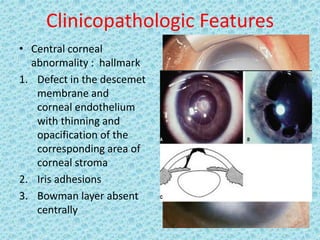








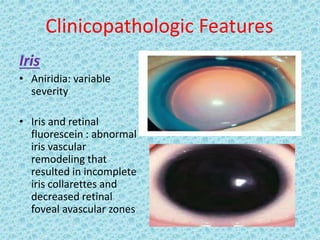

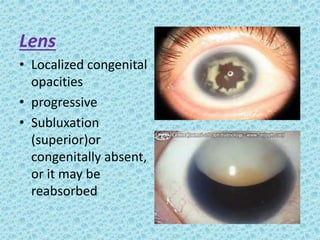









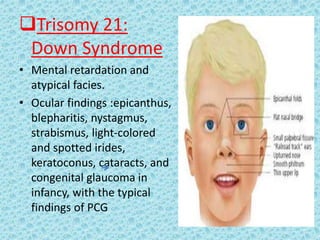







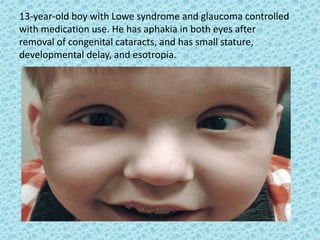







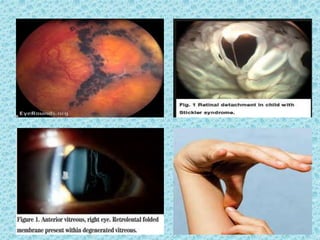


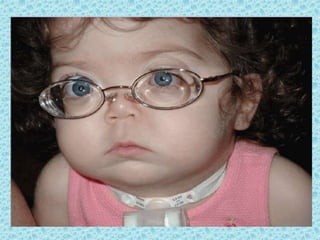



The document discusses developmental glaucomas associated with various anterior segment dysgenesis forms, including conditions like Axenfeld-Rieger syndrome, Peters anomaly, and aniridia, detailing their ocular and systemic features. It highlights genetic links, inheritance patterns, and differential diagnoses for these conditions, as well as common ocular abnormalities and associated systemic anomalies. The work emphasizes the high incidence of glaucoma in these syndromes and outlines potential mechanisms relating to abnormalities in the trabecular meshwork and other ocular structures.























































































