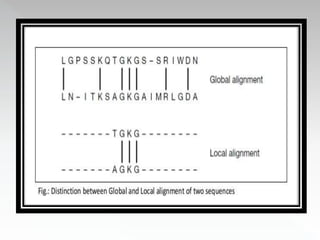
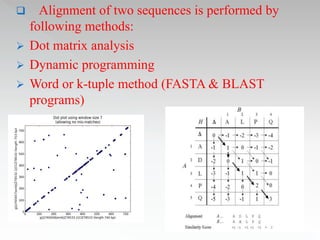
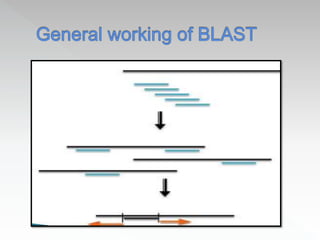
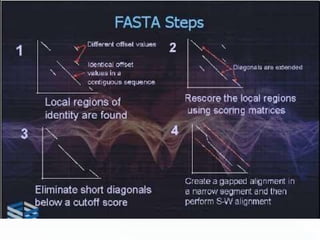
This document discusses sequence alignment methods. It describes global and local alignment, and algorithms used for alignment including dot matrix analysis, dynamic programming, and word/k-tuple methods as implemented in FASTA and BLAST programs. BLAST and FASTA are described as popular tools for sequence database searches that use heuristic methods and word matching to quickly identify regions of local similarity.