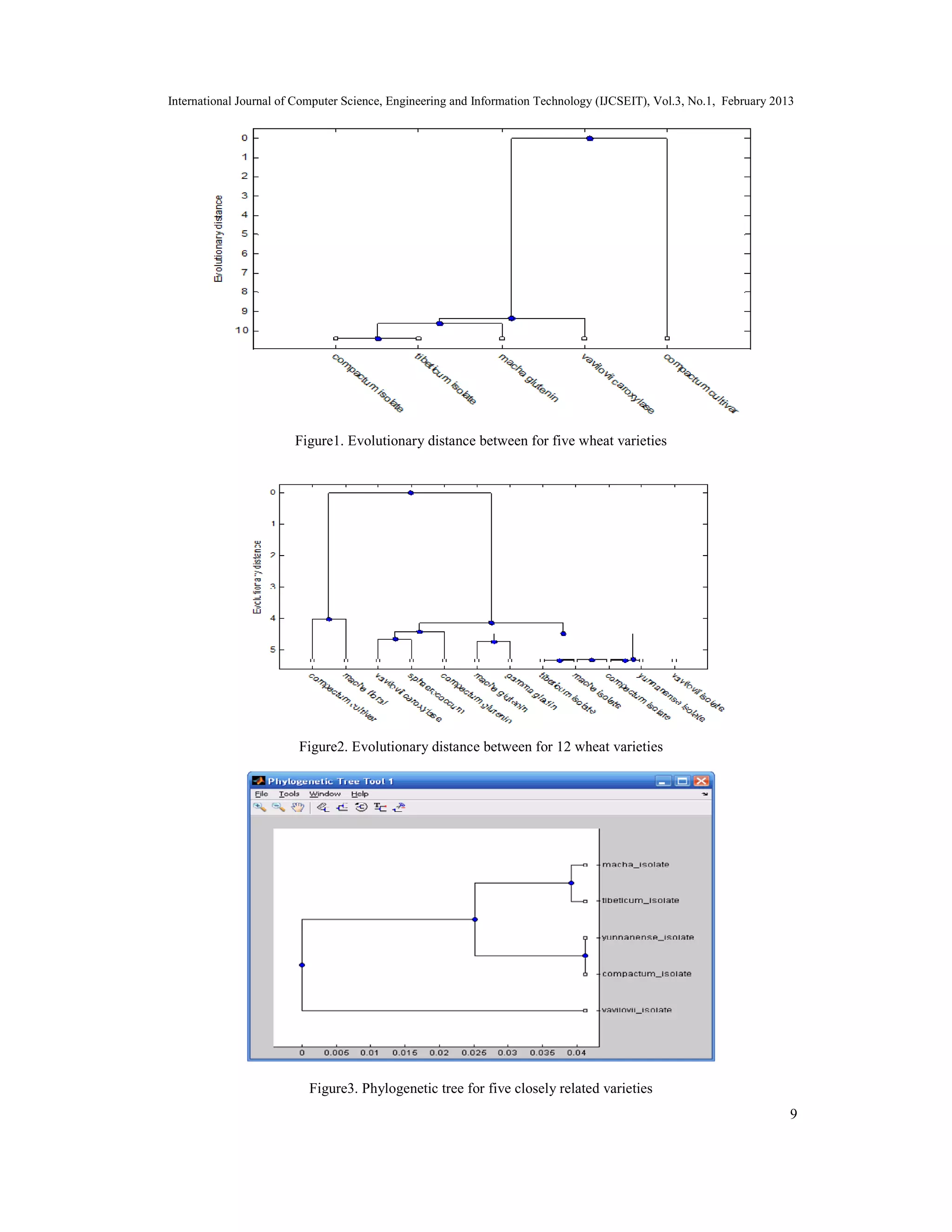
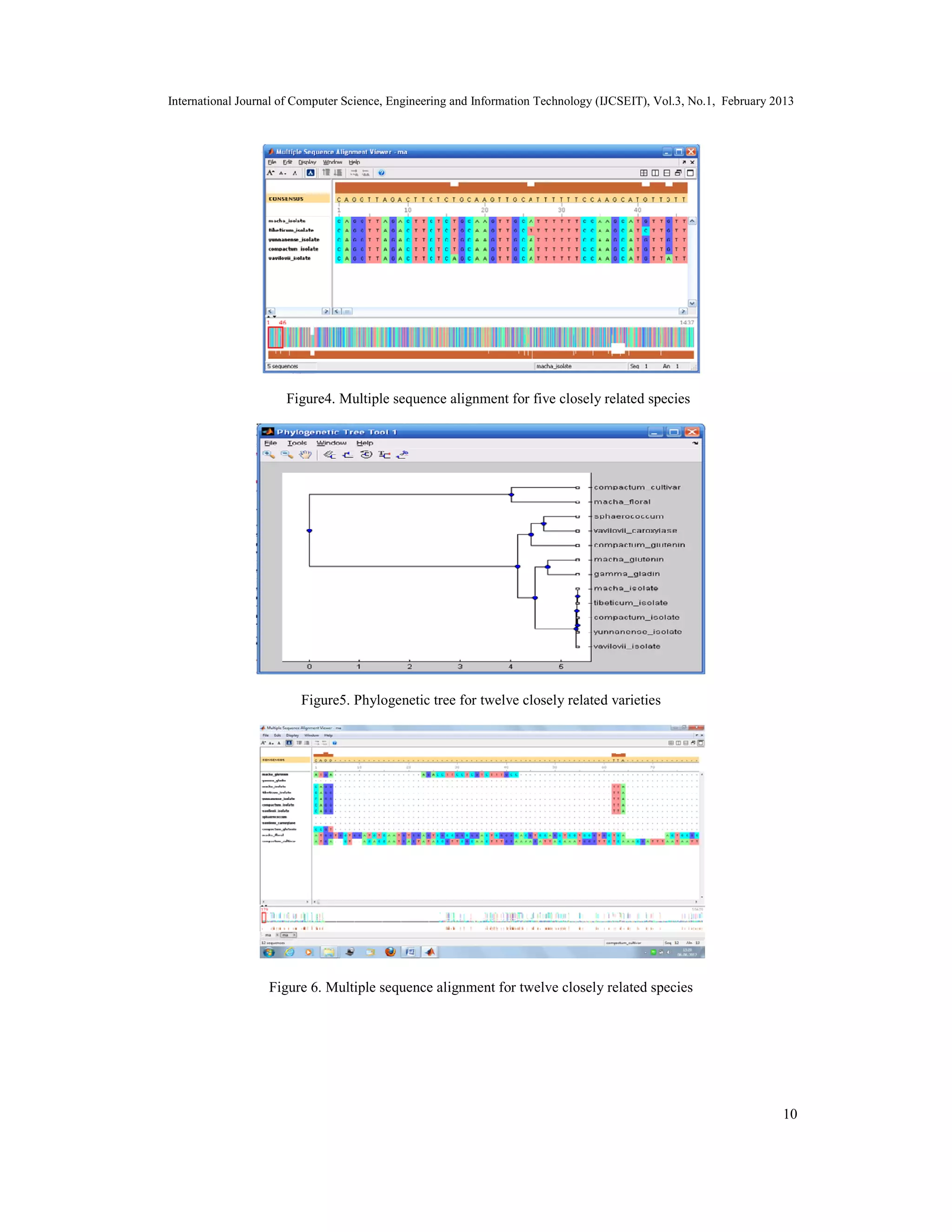
The document discusses the implementation of a hierarchical clustering method for multiple sequence alignment and phylogenetic tree construction, particularly using Triticum wheat varieties from the NCBI databank. It details various alignment methods, data mining techniques, and the methodology for calculating evolutionary distances using the Jukes-Cantor model, culminating in the construction of phylogenetic trees. The study concludes by addressing the complexity of multiple sequence alignment and presents the aligned sequences as a solution.

![International Journal of Computer Science, Engineering and Information Technology (IJCSEIT), Vol.3, No.1, February 2013
2
columns. Mismatches can be interpreted, if two sequences in an alignment share a common
ancestor and gaps as indels i.e. insertion or deletion mutations introduced in lineages. In DNA
sequence alignment, the degree of similarity between amino acids occupying a particular position
in the sequence can be presented as a rough measure of how conserved a particular region is
among lineages. The substitutions of nucleotides whose side chains have similar biochemical
properties suggest that this region has structural or functional importance.
2. DATA MINING
Data mining is the process of extracting patterns from data. Data mining is becoming an
increasingly important tool to transform this data into information. As part of the larger process
known as knowledge discovery, data mining is the process of extracting information from large
volumes of data. This is achieved through the identification and analysis of relationships and
trends within commercial databases. Data mining is used in areas as diverse as space exploration
and medical research. Data mining tools predict future trends and behaviors, allowing businesses
to make proactive, knowledge-driven decisions. The knowledge discovery process includes:
• Data Cleaning
• Data integration
• Data selection
• Data Transformation
• Data Mining
• Pattern Evaluation
• Knowledge Presentation
2.1 Data Mining Techniques
To design the data mining model the choice has to be made from various data mining techniques
[6][8], which are as follows:
a. Cluster Analysis
• Hierarchical Analysis
• Non- Hierarchical Analysis
b. Outlier Analysis
c. Induction
d. Online Analytical Processing (OLAP)
e. Neural Networks
f. Genetic Algorithms
g. Deviation Detection
h. Support Vector Machines
i. Data Visualization
j. Sequence Mining
In the present work we have adopted Hierarchical Cluster Analysis as a Data Mining approach, as
it is most suitable to work for a common group of protein sequences.
2.2 Data Mining Methods
Prediction and description are two important goals of data mining. Prediction uses the current
database to predict unknown or probable values of other variables of interest, and description
extracts the human-interpretable patterns describing the data.](https://image.slidesharecdn.com/3113ijcseit01-180327100205/75/International-Journal-of-Computer-Science-Engineering-and-Information-Technology-IJCSEIT-2-2048.jpg)







![International Journal of Computer Science, Engineering and Information Technology (IJCSEIT), Vol.3, No.1, February 2013
12
• The model can be extended for protein sequence alignment.
• Various Scoring matrices like PAM and BLOSUM can be incorporated for computing the
evolutionary distances.
8. REFERENCES
[1] B.Bergeron, Bioinformatics Computing, Pearson Education, 2003, pp. 110- 160.
[2] Clare, A. Machine learning and data mining for yeast functional genomics Ph.D. thesis, University of
Wales, 2003.
[3] Han, J. and Kamber M., Data Mining: Concepts and Techniques, Morga Kaufmann Publishers, 2004,
pp. 19-25.
[4] Jiang, D. Tang, C. and Zhang, A., “Cluster Analysis for Gene Expression Data”, IEEE Transactions
on knowledge and data engineering, vol. 11, 2004, pp. 1370-1386.
[5] Kai, L. and Li-Juan, C., “Study of Clustering Algorithm Based on Model Data”, International
Conference on Machine Learning and Cybernetics, Honkong, 2007, Volume 7, No. 2., 3961-3964
[6] Kantardzic, M., Data Mining: Concepts, Models, Methods, and Algorithms, John Wiley & Sons,
2000, pp. 112-129.
[7] Baxevanis, A.D and Ouellette, B.(2001) ” Sequence alignment and Database Searching” in
Bioinformatics: A Practical Guide to the Analysis of Genes and Proteins. John Wiley & Sons, New
York, pp. 187-212.
[8] Tzeng YH. Et al. (2004) “Comparison of three methods for estimating rates of synonymous and
nonsynonymous nucleotide substitutions”, Molecular. Biology Evol. , pp. 2290-8.
AUTHOR
Harmandeep Singh has received B-Tech degree from Punjab Technical University,
Jalandhar in 2009 and his M-Tech Degree from Punjab Technical University, Jalandhar
in 2012.
Er. Rajbir Singh Cheema is working as a Head of Department in Lala Lajpat Rai
Institute of Engineering & Technology, Moga, Punjab. His research interests are in the
fields of Data Mining, Open Reading Frame in Bioinformatics, Gene Expression
Omnibus, Cloud Computing, Routing Algorithms, Routing Protocols Load Balancing
and Network Security. He has published many national and international papers.
Navjot Kaur has received B-Tech degree from Punjab Technical University, Jalandhar in 2010 and
pursuing her M-Tech Degree from Punjab Technical University, Jalandhar. She is working as an Assitant
Professor in Lala Lajpat Rai Institute of Engineering & Technology, Moga, Punjab.](https://image.slidesharecdn.com/3113ijcseit01-180327100205/75/International-Journal-of-Computer-Science-Engineering-and-Information-Technology-IJCSEIT-12-2048.jpg)






















































































