Download as PDF, PPTX



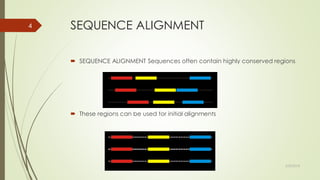










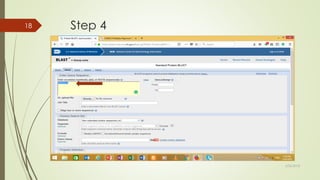
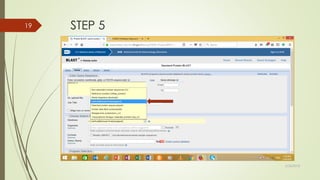
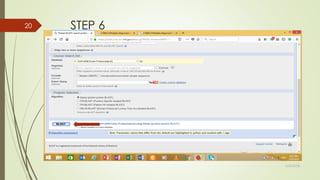

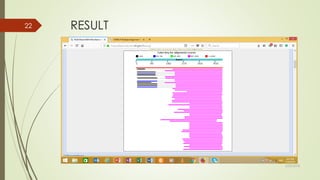








This document discusses multiple sequence alignment tools. It describes how multiple sequence alignments (MSAs) are used to identify conserved regions across related sequences and determine consensus sequences. The document outlines different types of MSA, including progressive and iterative methods. It also describes the steps to perform a multiple sequence alignment using COBALT at NCBI, including uploading sequences, running the alignment, and editing results. The progressive method is noted as the most widely used due to its speed and accuracy, though it may not produce globally optimal alignments.



































































