The document presents an overview of molecular docking using AutoDock tools, explaining the computational process for predicting favorable ligand conformations within macromolecule binding sites. It details prerequisites such as 3D structures of proteins and ligands, docking methods (rigid and flexible), and components involved in the docking process, including search algorithms and scoring functions. Furthermore, the document includes step-by-step instructions for setting up macromolecules and ligands, configuring docking parameters, and analyzing AutoDock results.
Overview
Molecular docking isa computational
process which involves the prediction of
favorable conformation of a ligand within
the binding site of a target macromolecule.
• 3D structure of the macromolecule
Target Structure
• Information about the location of the active site of target
Binding Site
• 3D structure of the compounds whose binding is to be studied
Ligand
The prerequisites for molecular docking:
3.
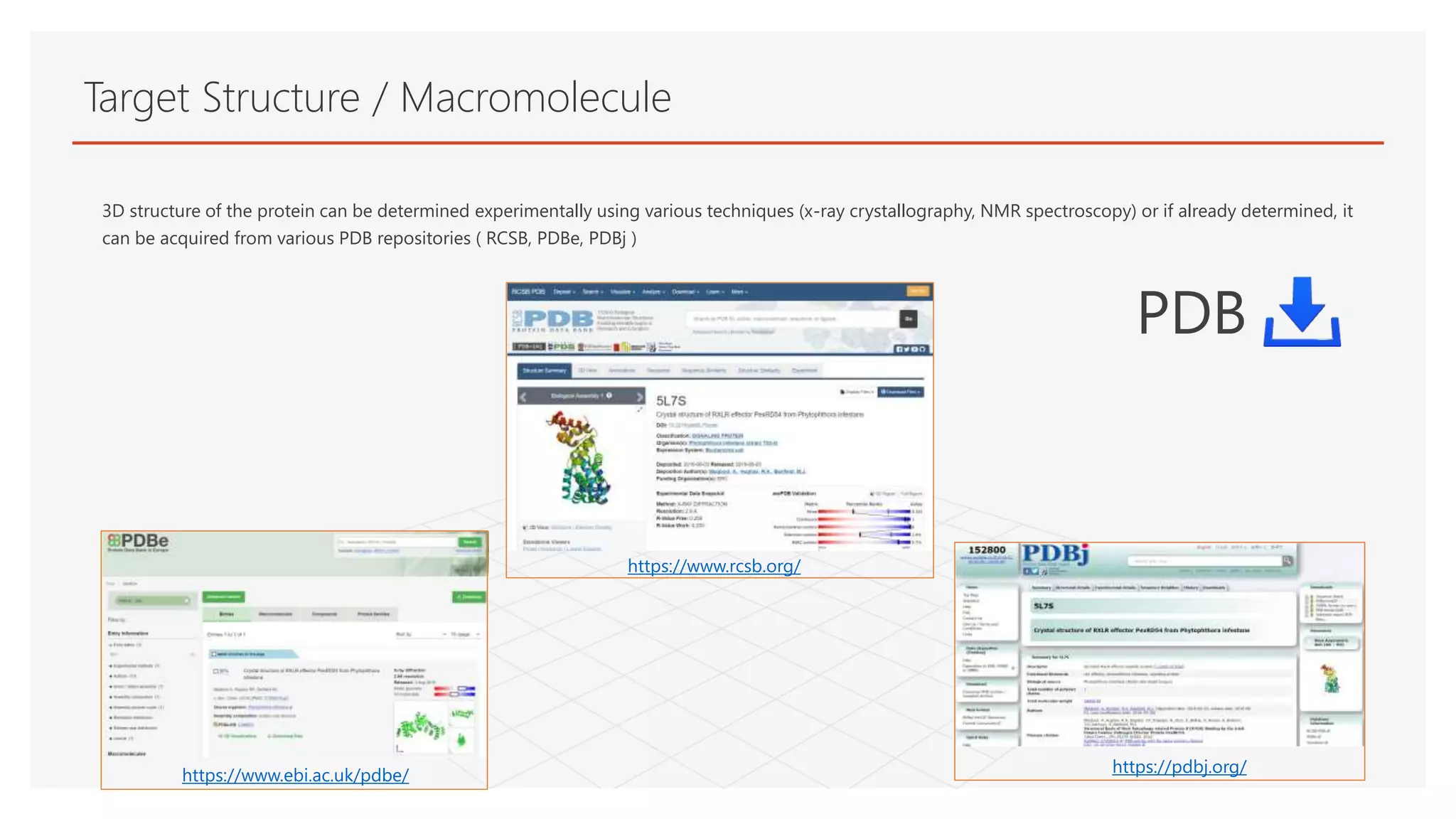
Target Structure /Macromolecule
3D structure of the protein can be determined experimentally using various techniques (x-ray crystallography, NMR spectroscopy) or if already determined, it
can be acquired from various PDB repositories ( RCSB, PDBe, PDBj )
https://www.rcsb.org/
https://www.ebi.ac.uk/pdbe/ https://pdbj.org/
PDB
4.
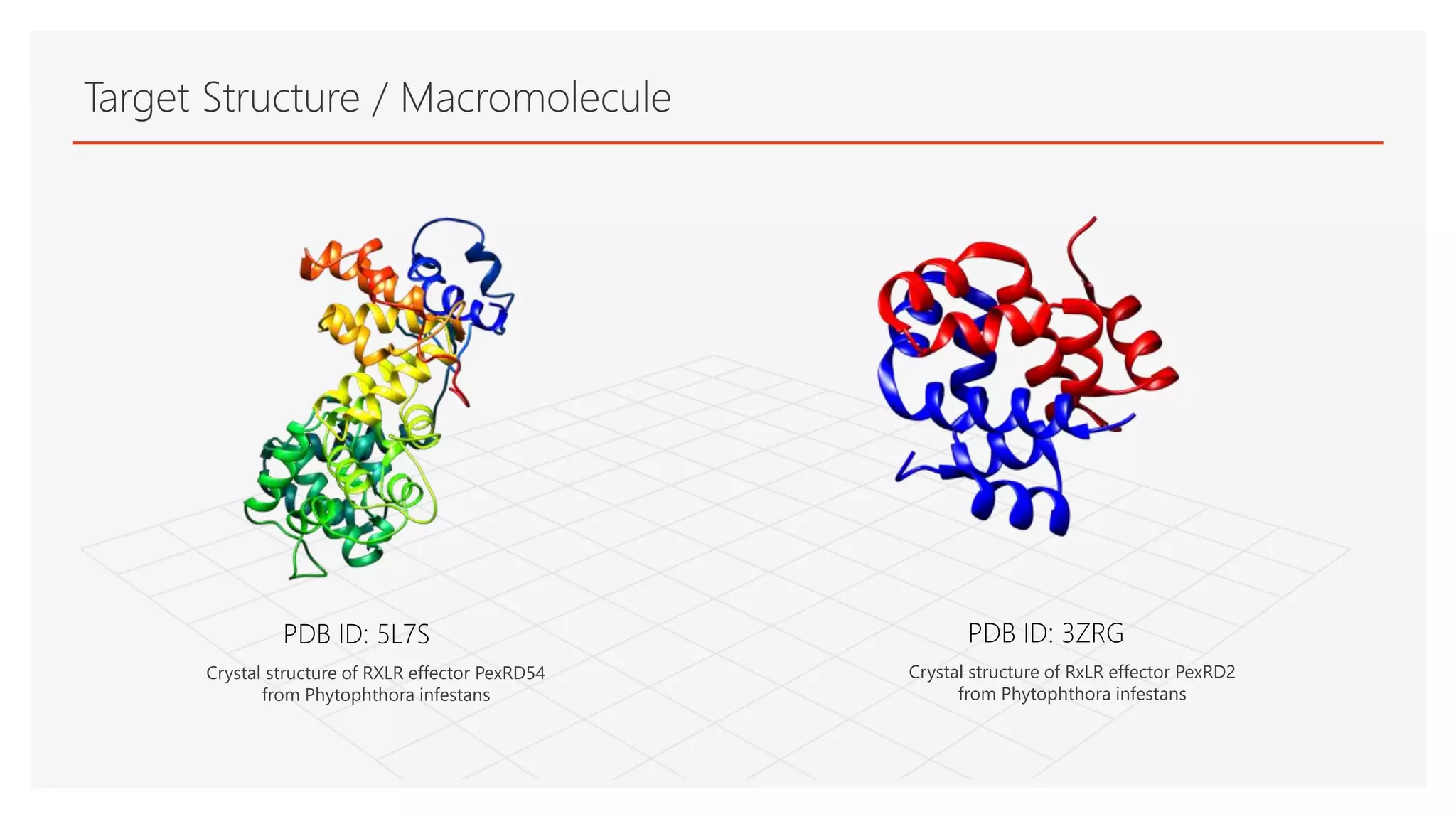
Target Structure /Macromolecule
PDB ID: 5L7S PDB ID: 3ZRG
Crystal structure of RxLR effector PexRD2
from Phytophthora infestans
Crystal structure of RXLR effector PexRD54
from Phytophthora infestans
5.
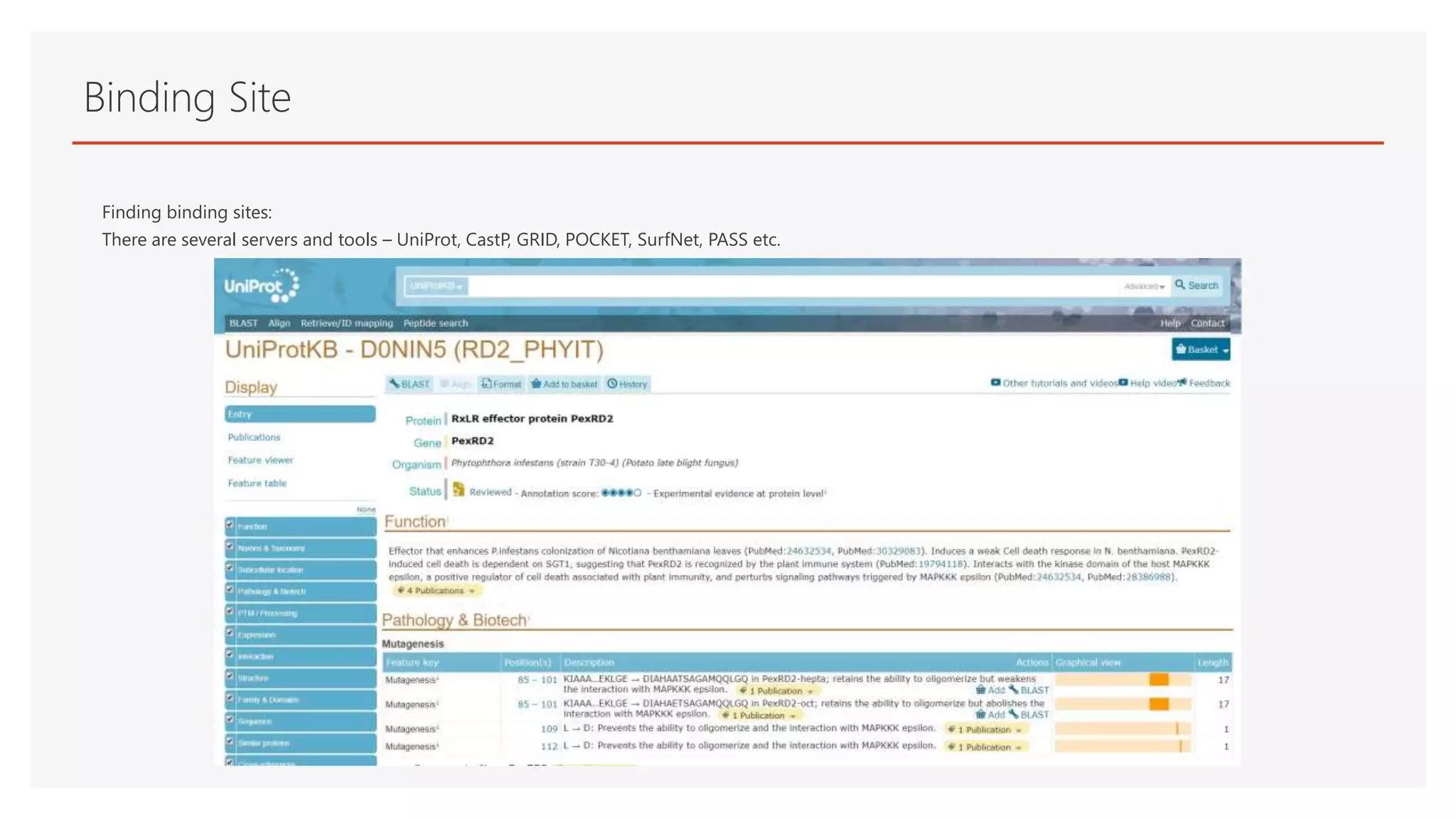
Binding Site
Finding bindingsites:
There are several servers and tools – UniProt, CastP, GRID, POCKET, SurfNet, PASS etc.
6.
Ligand
• Filtered FASTAsequence
• Structure prediction in QUARK ----> 3D structure (pdb)
• Structure Validation in PSVS
*Small peptide sequence ID: 0489
* this peptide is translated from non coding DNA ( junk DNA ) and modelled into 3D structure by
ab-inito approach [Ref: Dr. Pawan Dhar, Synthetic Biology Lab, School of Biotechnology, JNU]
Rigid and FlexibleDocking
DOCKING
RIGID BODY
DOCKING
FLEXIBLE DOCKING
The rigid docking methods do not consider ligand and receptor flexibility.
- Orientation is taken as a whole
- No freedom of rotation in their side change
The flexible docking ligand and target molecules both are allowed to change
conformations during the docking process
- More reliable
- Requires more computational power
9.
Components in DockingProcess
Docking programs are generally devised as combination of two components:
A search algorithm and scoring function.
1. Search Algorithms – identify the possible conformations
2. Scoring Functions – prediction of interaction energy corresponding to each of the predicted binding confirmation
DOCKING
Binding mode prediction
Binding affinity prediction
10.
Exhaustive
Conformational
Ensemble
Fragment Based
Search algorithmsplay a crucial role in identifying all the conformations that are feasible in the search space
Search Algorithms
Search Algorithm
Shape Matching Systematic Search Stochastic Algorithm
Ligand divided in fragments
Fragments docked individually into the target
docked fragments linked covalently
Address ligand flexibility problem
Use ensemble of ligand
conformations previously generated
through rigid docking
Various conformations generated by
energy sampling over the entire range of
degrees of freedom & all global
& local minima visited
Genetic Algorithm
11.
Genetic Search Algorithm,a stochastic search method
It applies theories of evolution and natural selection.
An initial population of solutions is created through genetic operators (mutations, crossovers and migrations), and ranked using the survival of
the fittest.
The initial population covers a wide area of the energy landscape.
The lowest energy conformations are selected as templates for the generation of the next population.
GA requires generation of an initial population of ligand conformations.
Search Algorithms
12.
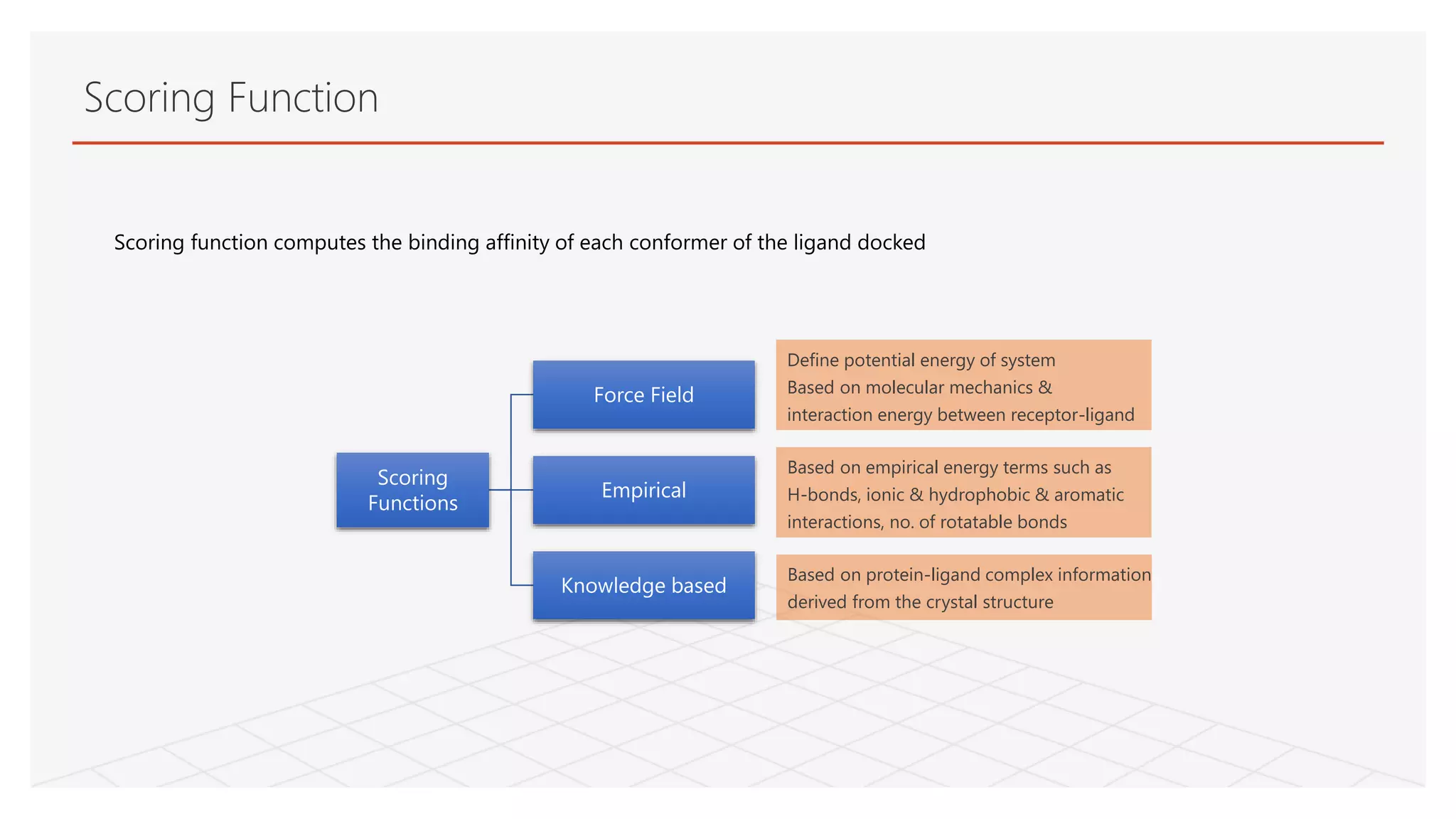
Scoring function computesthe binding affinity of each conformer of the ligand docked
Scoring Function
Scoring
Functions
Force Field
Empirical
Knowledge based
Define potential energy of system
Based on molecular mechanics &
interaction energy between receptor-ligand
Based on empirical energy terms such as
H-bonds, ionic & hydrophobic & aromatic
interactions, no. of rotatable bonds
Based on protein-ligand complex information
derived from the crystal structure
13.
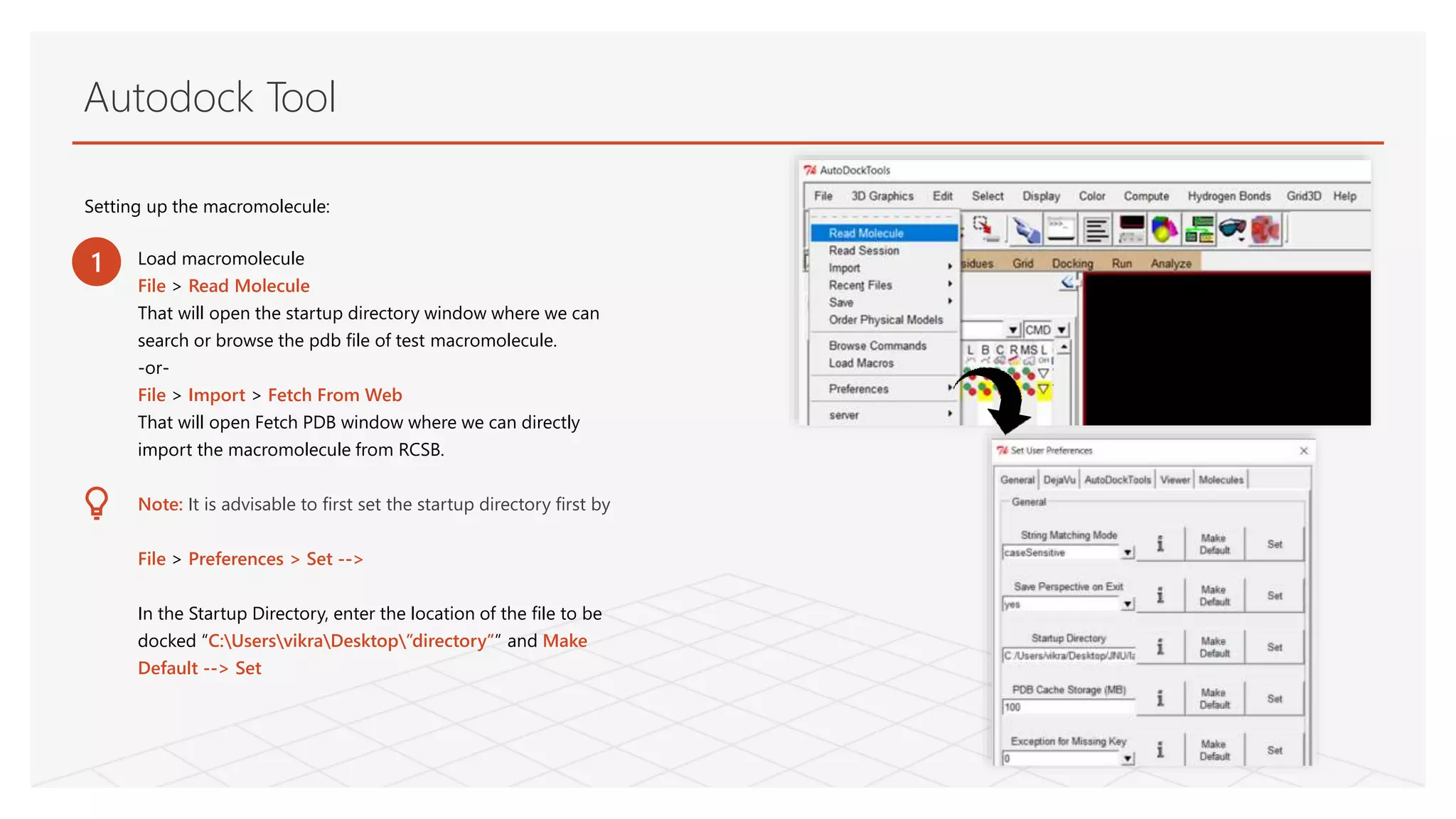
Autodock Tool
Setting upthe macromolecule:
Load macromolecule
File > Read Molecule
That will open the startup directory window where we can
search or browse the pdb file of test macromolecule.
-or-
File > Import > Fetch From Web
That will open Fetch PDB window where we can directly
import the macromolecule from RCSB.
Note: It is advisable to first set the startup directory first by
File > Preferences > Set -->
In the Startup Directory, enter the location of the file to be
docked “C:UsersvikraDesktop”directory”” and Make
Default --> Set
1
14.
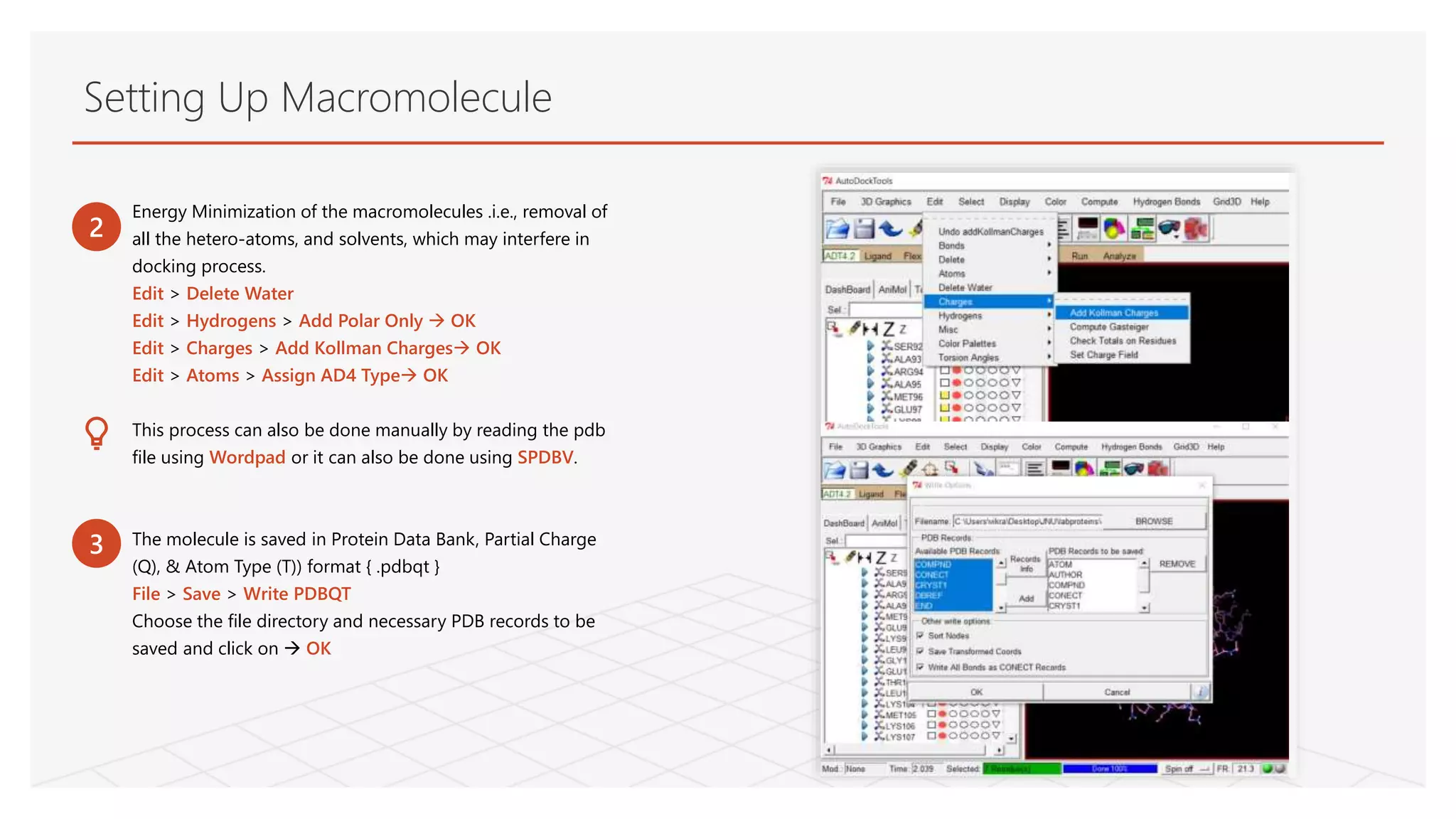
Energy Minimization ofthe macromolecules .i.e., removal of
all the hetero-atoms, and solvents, which may interfere in
docking process.
Edit > Delete Water
Edit > Hydrogens > Add Polar Only OK
Edit > Charges > Add Kollman Charges OK
Edit > Atoms > Assign AD4 Type OK
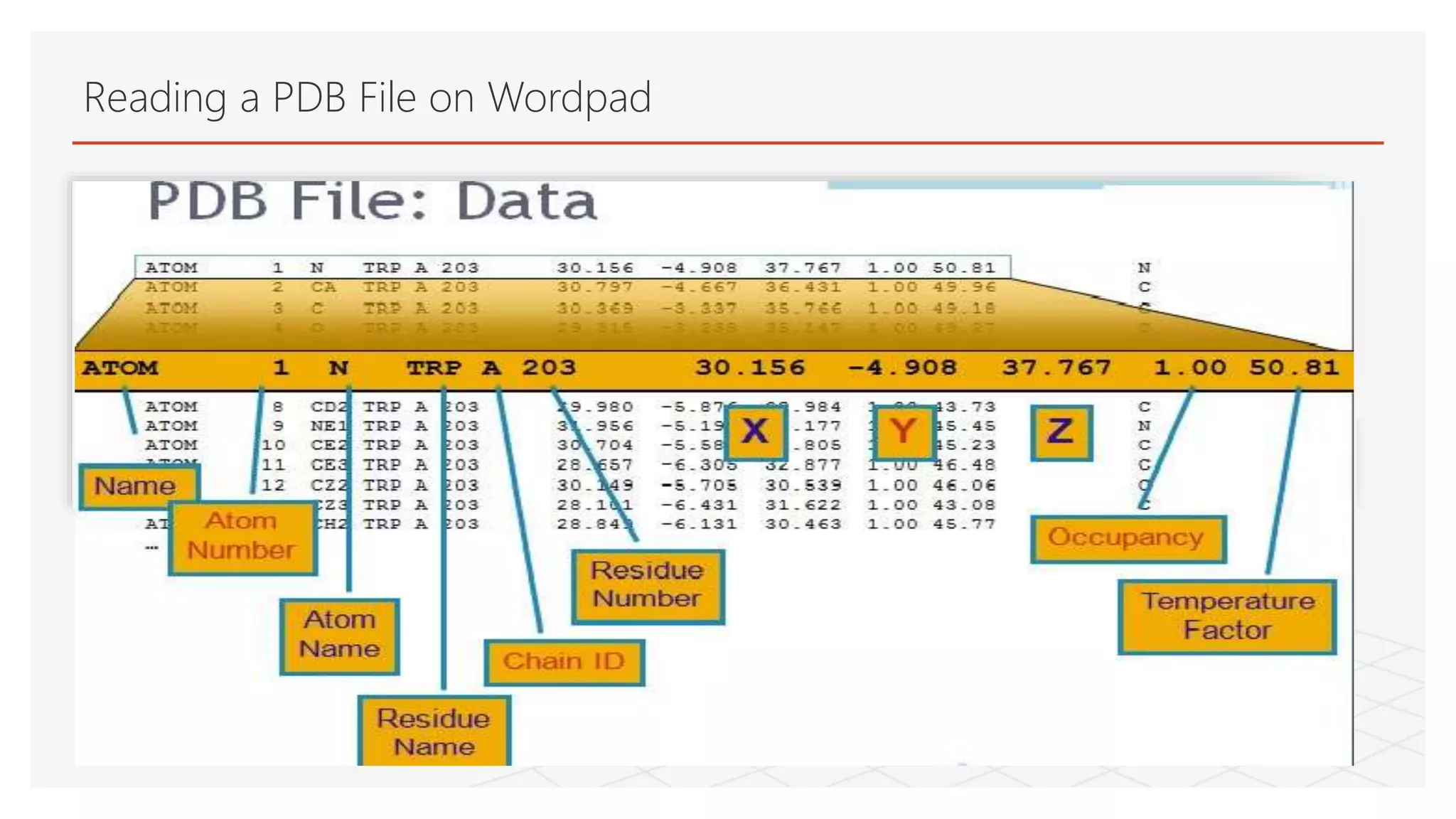
This process can also be done manually by reading the pdb
file using Wordpad or it can also be done using SPDBV.
The molecule is saved in Protein Data Bank, Partial Charge
(Q), & Atom Type (T)) format { .pdbqt }
File > Save > Write PDBQT
Choose the file directory and necessary PDB records to be
saved and click on OK
2
3
Setting Up Macromolecule
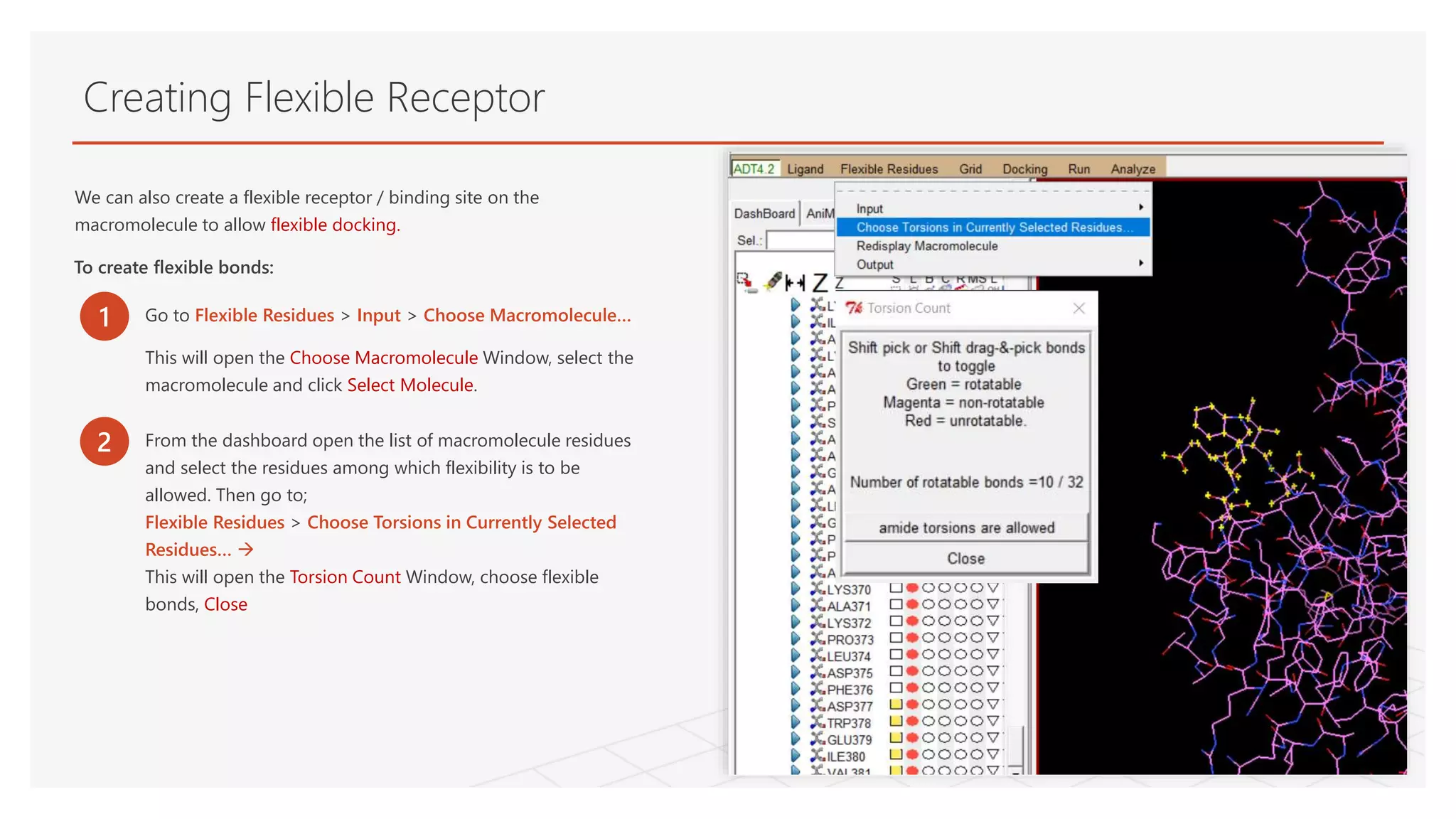
Creating Flexible Receptor
1Go to Flexible Residues > Input > Choose Macromolecule…
This will open the Choose Macromolecule Window, select the
macromolecule and click Select Molecule.
We can also create a flexible receptor / binding site on the
macromolecule to allow flexible docking.
To create flexible bonds:
2 From the dashboard open the list of macromolecule residues
and select the residues among which flexibility is to be
allowed. Then go to;
Flexible Residues > Choose Torsions in Currently Selected
Residues…
This will open the Torsion Count Window, choose flexible
bonds, Close
17.
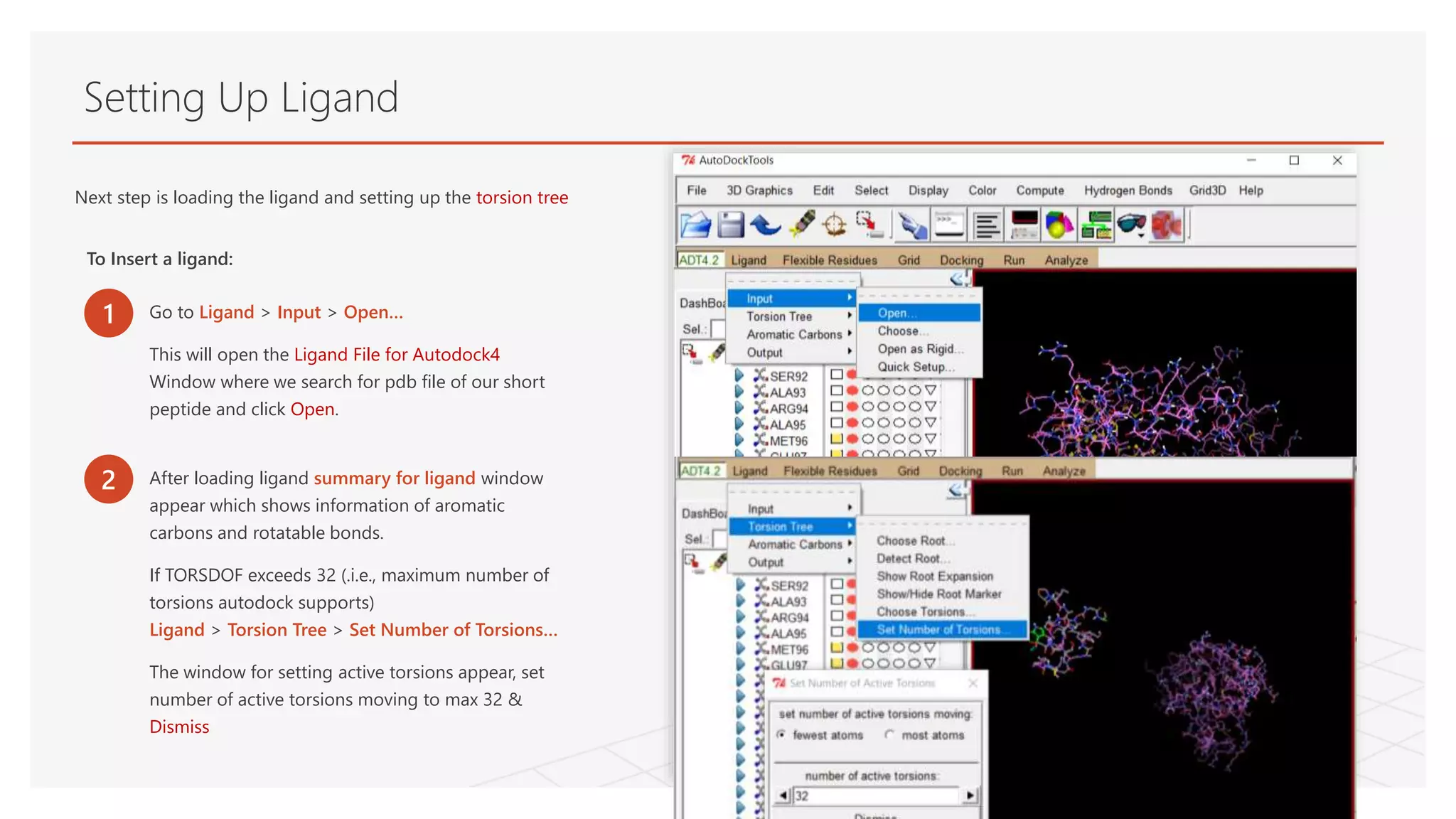
Setting Up Ligand
Nextstep is loading the ligand and setting up the torsion tree
To Insert a ligand:
1 Go to Ligand > Input > Open…
This will open the Ligand File for Autodock4
Window where we search for pdb file of our short
peptide and click Open.
2 After loading ligand summary for ligand window
appear which shows information of aromatic
carbons and rotatable bonds.
If TORSDOF exceeds 32 (.i.e., maximum number of
torsions autodock supports)
Ligand > Torsion Tree > Set Number of Torsions…
The window for setting active torsions appear, set
number of active torsions moving to max 32 &
Dismiss
18.
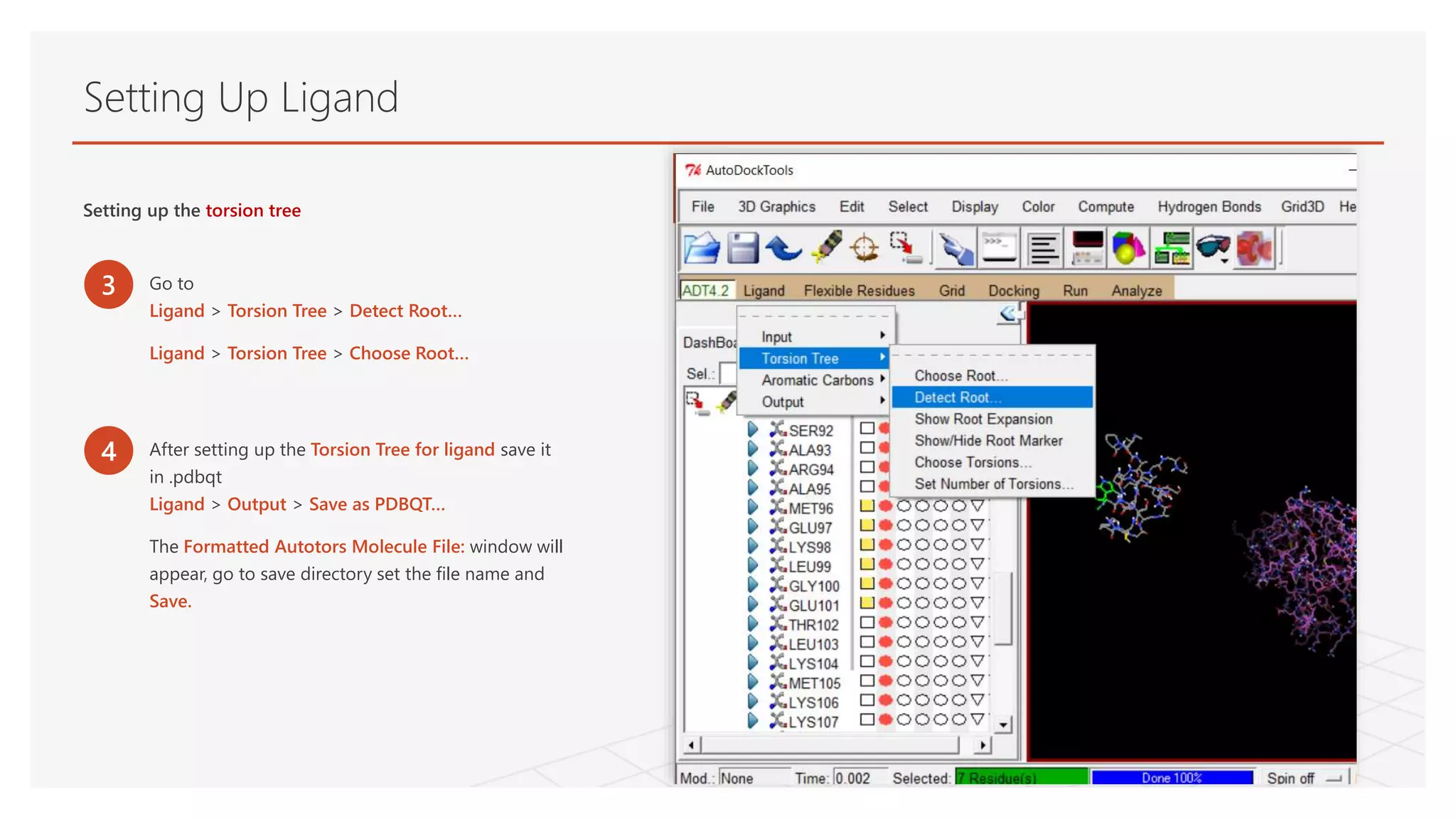
Setting Up Ligand
Settingup the torsion tree
3 Go to
Ligand > Torsion Tree > Detect Root…
Ligand > Torsion Tree > Choose Root…
4 After setting up the Torsion Tree for ligand save it
in .pdbqt
Ligand > Output > Save as PDBQT…
The Formatted Autotors Molecule File: window will
appear, go to save directory set the file name and
Save.
19.
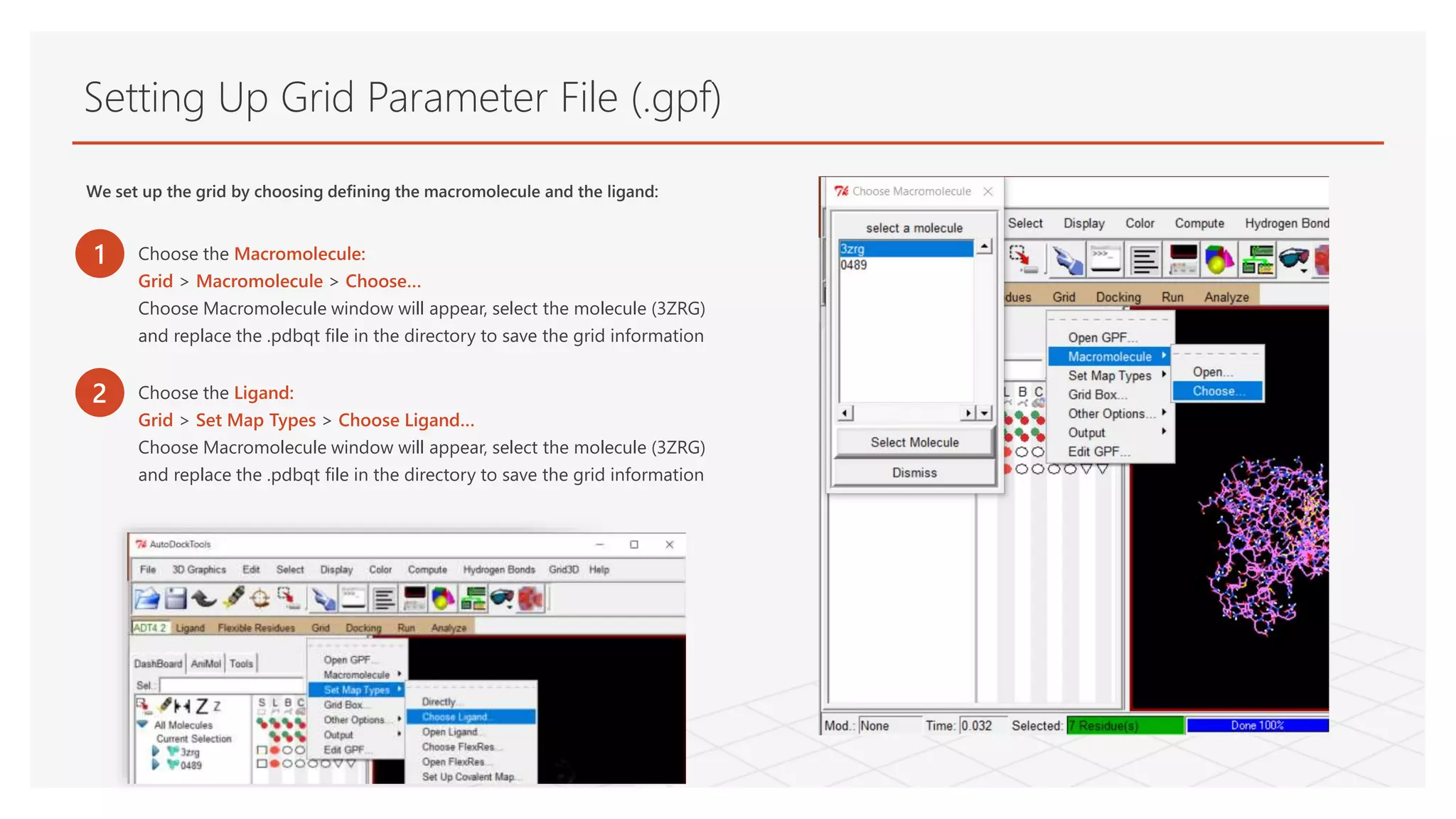
Setting Up GridParameter File (.gpf)
We set up the grid by choosing defining the macromolecule and the ligand:
1 Choose the Macromolecule:
Grid > Macromolecule > Choose…
Choose Macromolecule window will appear, select the molecule (3ZRG)
and replace the .pdbqt file in the directory to save the grid information
2 Choose the Ligand:
Grid > Set Map Types > Choose Ligand…
Choose Macromolecule window will appear, select the molecule (3ZRG)
and replace the .pdbqt file in the directory to save the grid information
20.
Setting Up GridParameter File (.gpf)
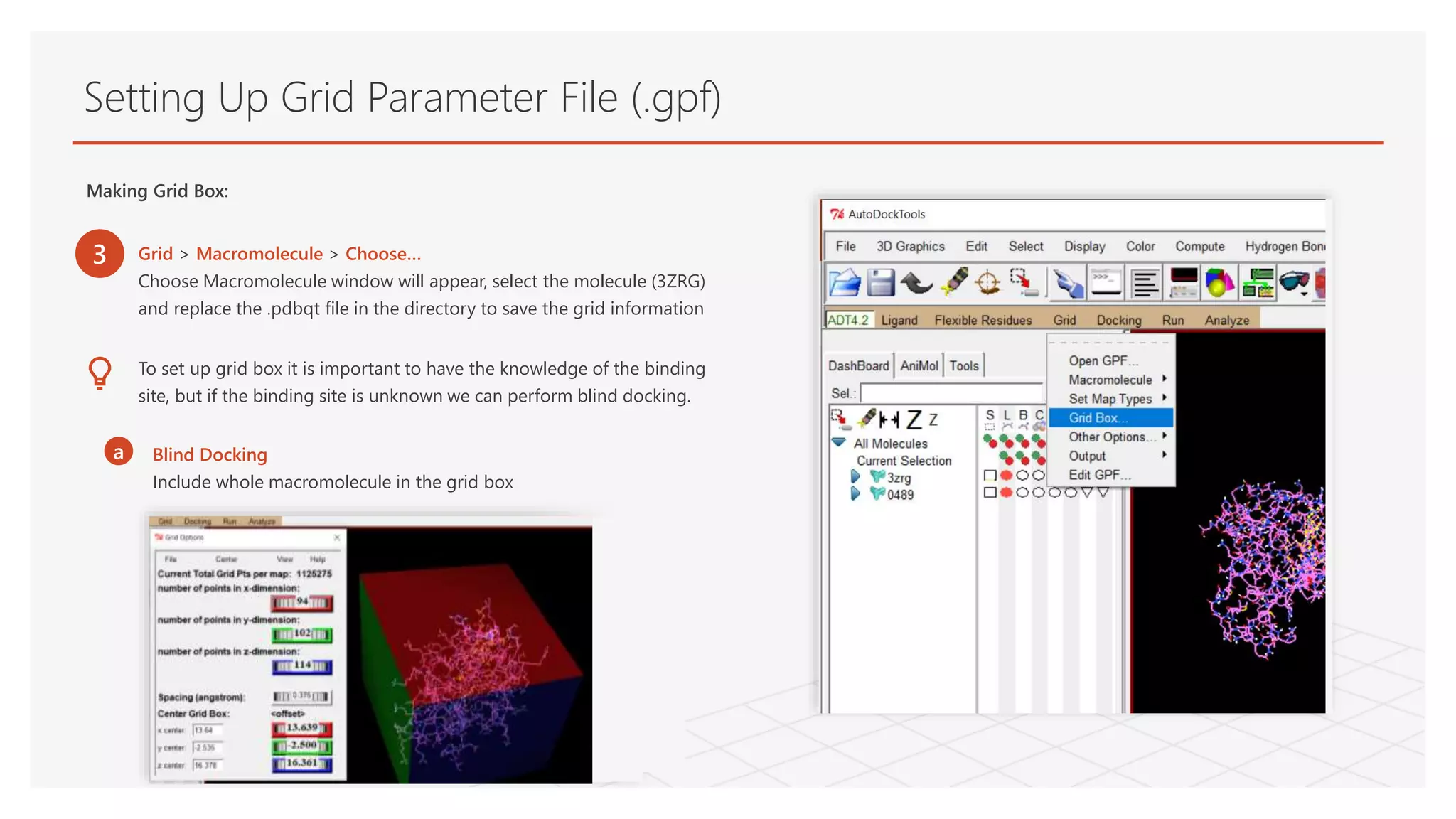
Making Grid Box:
3 Grid > Macromolecule > Choose…
Choose Macromolecule window will appear, select the molecule (3ZRG)
and replace the .pdbqt file in the directory to save the grid information
To set up grid box it is important to have the knowledge of the binding
site, but if the binding site is unknown we can perform blind docking.
a Blind Docking
Include whole macromolecule in the grid box
21.
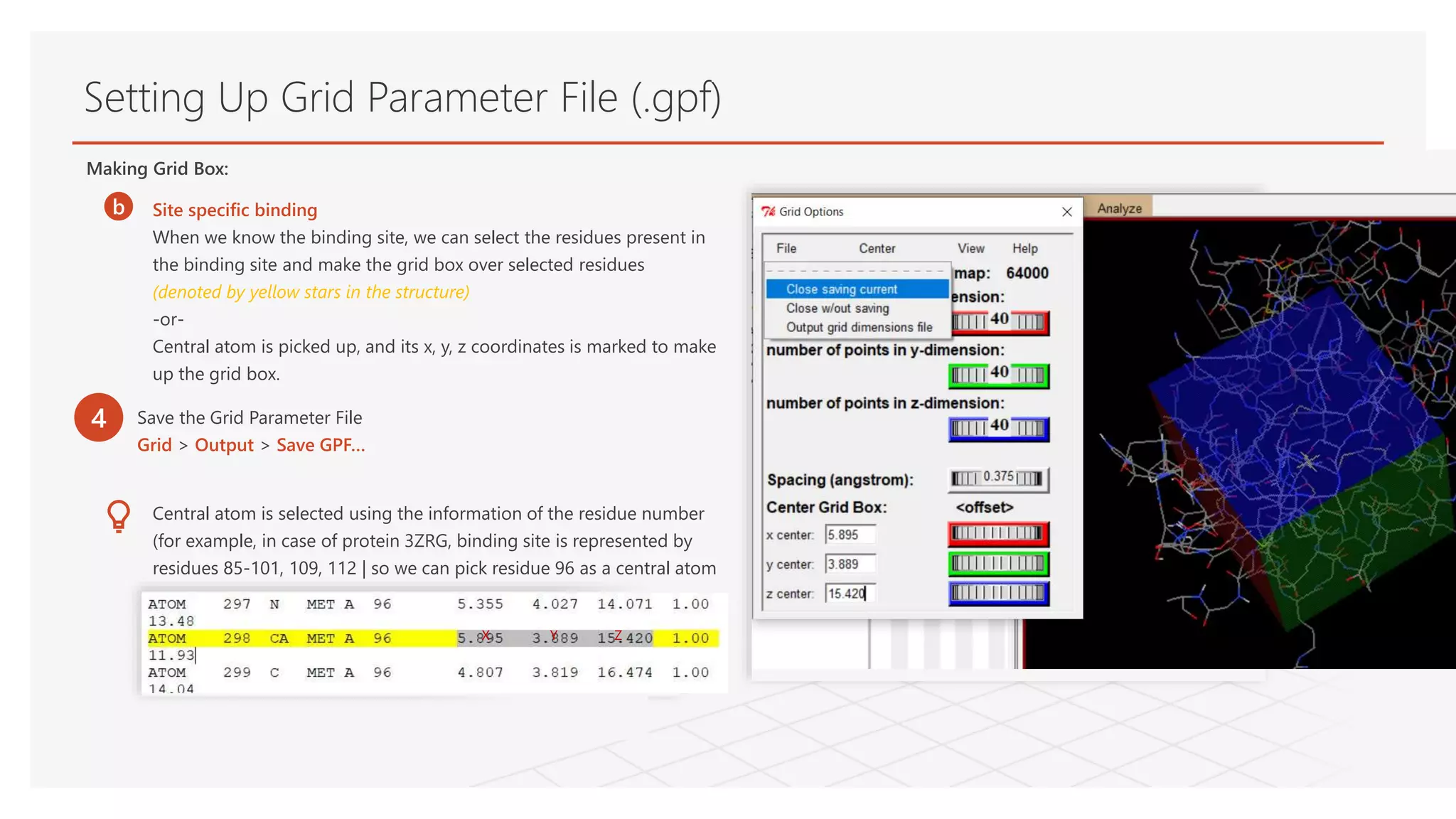
Setting Up GridParameter File (.gpf)
Making Grid Box:
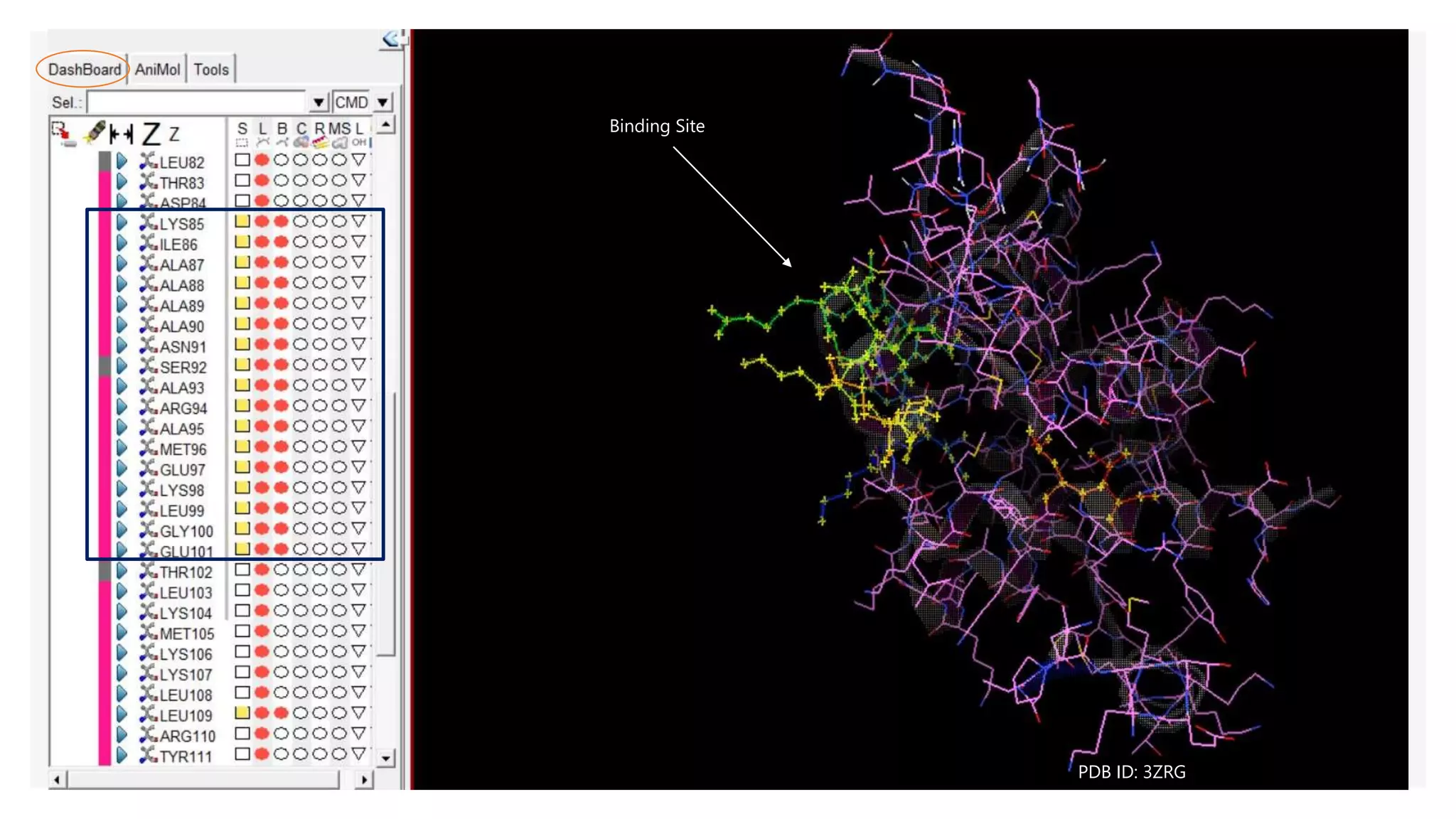
b Site specific binding
When we know the binding site, we can select the residues present in
the binding site and make the grid box over selected residues
(denoted by yellow stars in the structure)
-or-
Central atom is picked up, and its x, y, z coordinates is marked to make
up the grid box.
Central atom is selected using the information of the residue number
(for example, in case of protein 3ZRG, binding site is represented by
residues 85-101, 109, 112 | so we can pick residue 96 as a central atom
X Y Z
4 Save the Grid Parameter File
Grid > Output > Save GPF…
Generating Docking ParameterFile (.dpf)
To resize or crop your 3D model within a frame, you can use the pan and zoom tool.
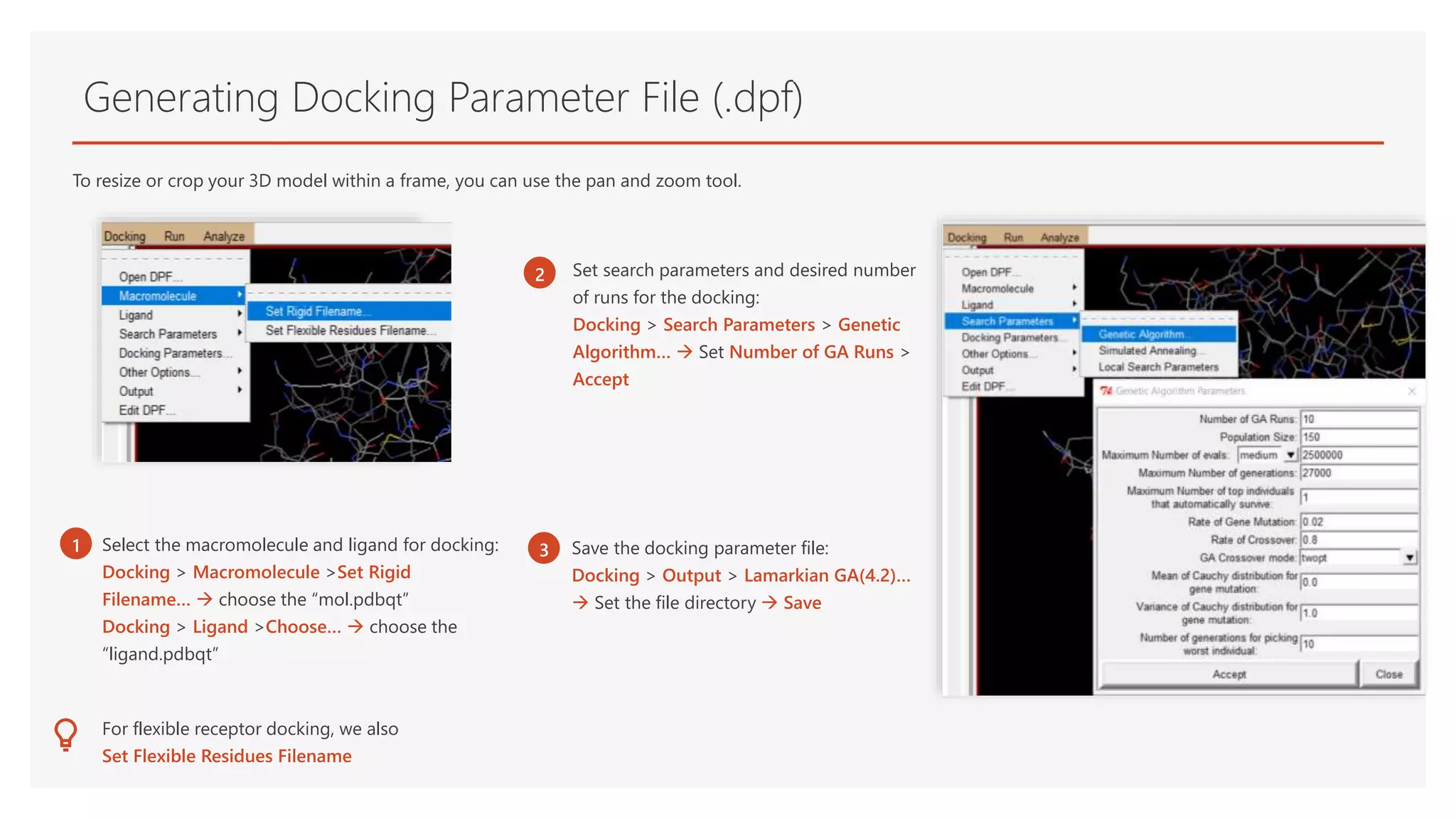
1 Select the macromolecule and ligand for docking:
Docking > Macromolecule >Set Rigid
Filename… choose the “mol.pdbqt”
Docking > Ligand >Choose… choose the
“ligand.pdbqt”
Set search parameters and desired number
of runs for the docking:
Docking > Search Parameters > Genetic
Algorithm… Set Number of GA Runs >
Accept
3 When you are finished editing, click
the Pan & Zoom button again to exit
Pan and Zoom mode.
For flexible receptor docking, we also
Set Flexible Residues Filename
Save the docking parameter file:
Docking > Output > Lamarkian GA(4.2)…
Set the file directory Save
2
3
24.
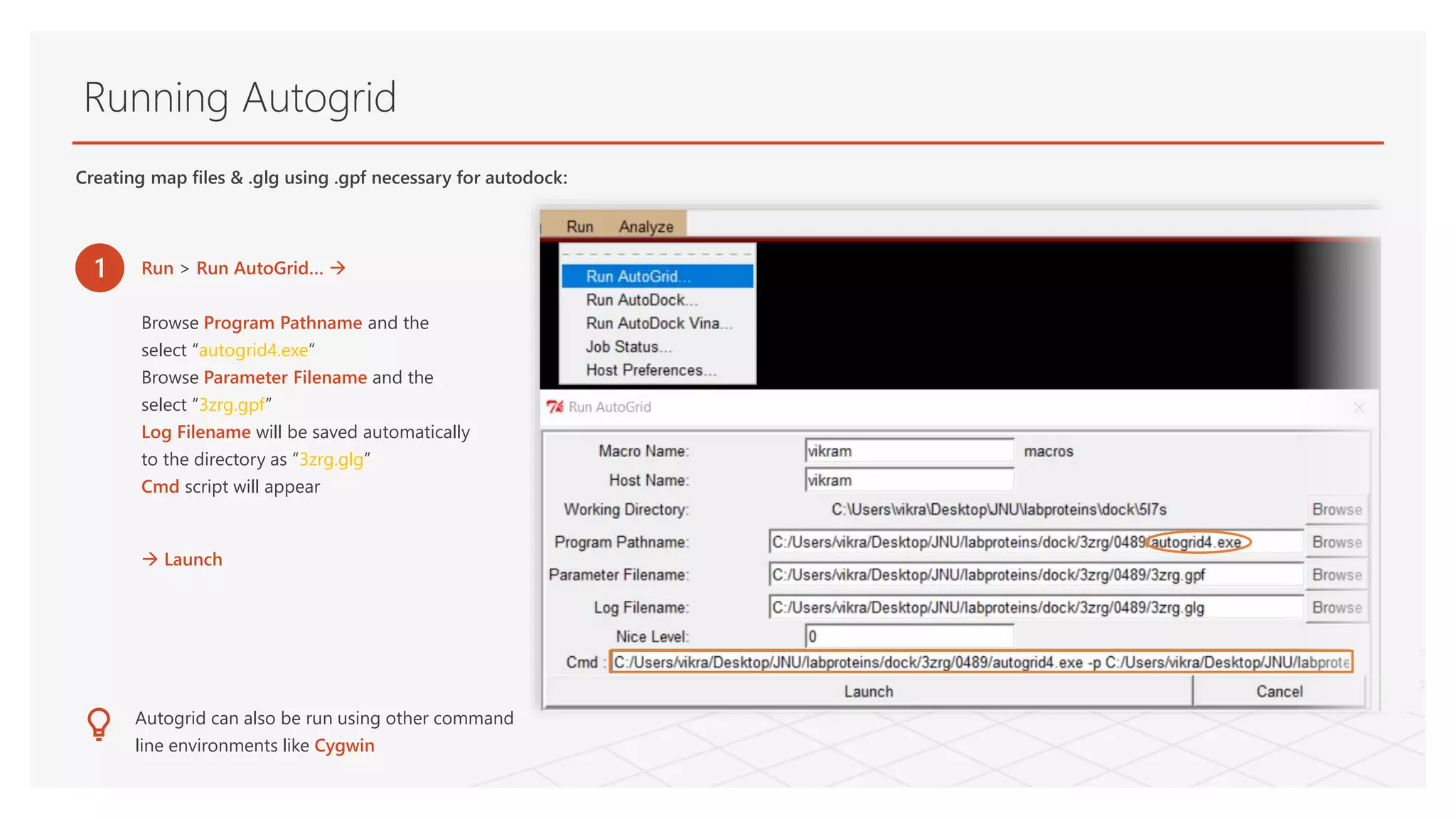
Running Autogrid
1 Run> Run AutoGrid…
Browse Program Pathname and the
select “autogrid4.exe”
Browse Parameter Filename and the
select “3zrg.gpf”
Log Filename will be saved automatically
to the directory as “3zrg.glg”
Cmd script will appear
Launch
Autogrid can also be run using other command
line environments like Cygwin
Creating map files & .glg using .gpf necessary for autodock:
25.
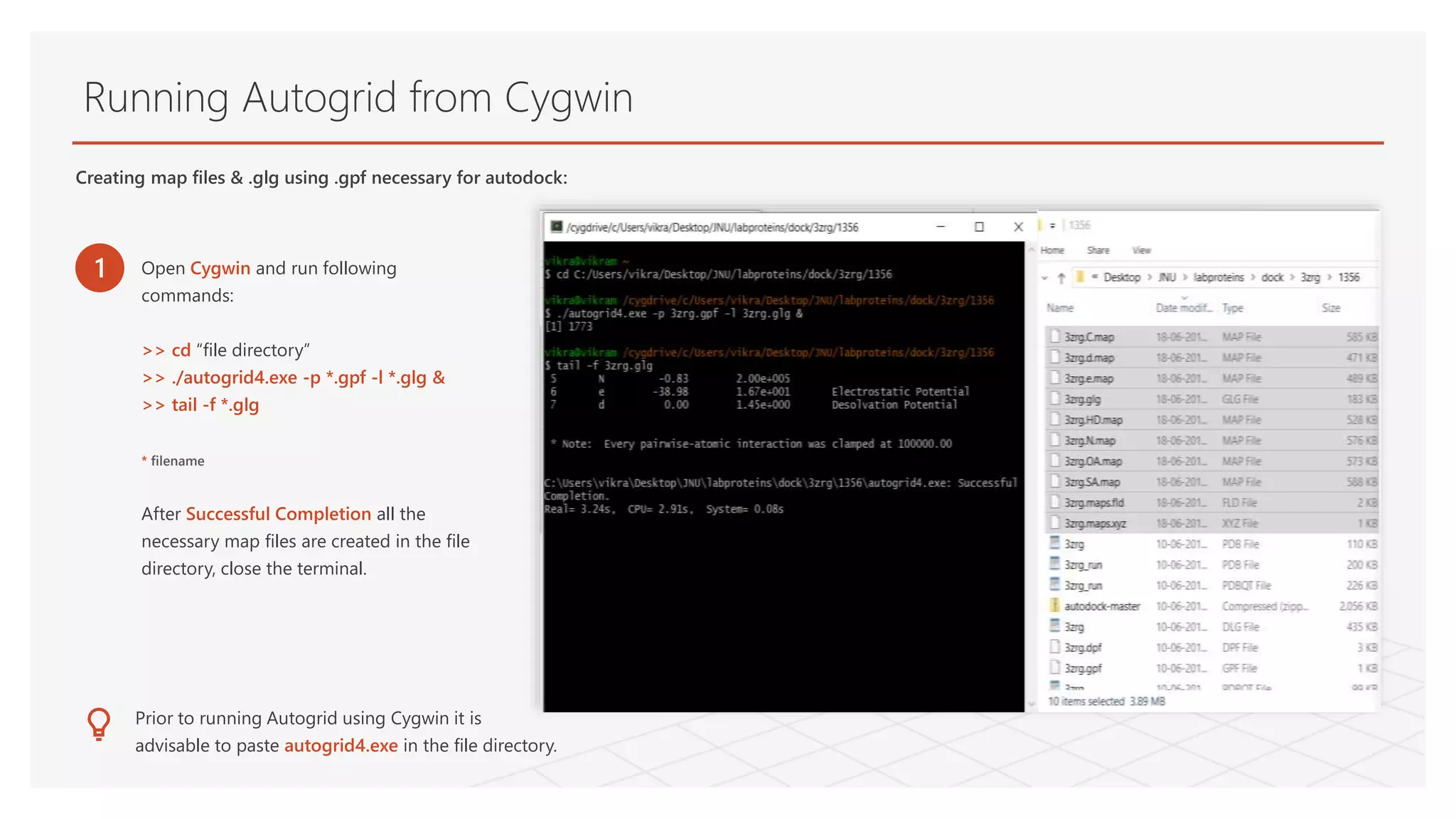
Running Autogrid fromCygwin
Creating map files & .glg using .gpf necessary for autodock:
1 Open Cygwin and run following
commands:
>> cd “file directory”
>> ./autogrid4.exe -p *.gpf -l *.glg &
>> tail -f *.glg
* filename
After Successful Completion all the
necessary map files are created in the file
directory, close the terminal.
Prior to running Autogrid using Cygwin it is
advisable to paste autogrid4.exe in the file directory.
26.
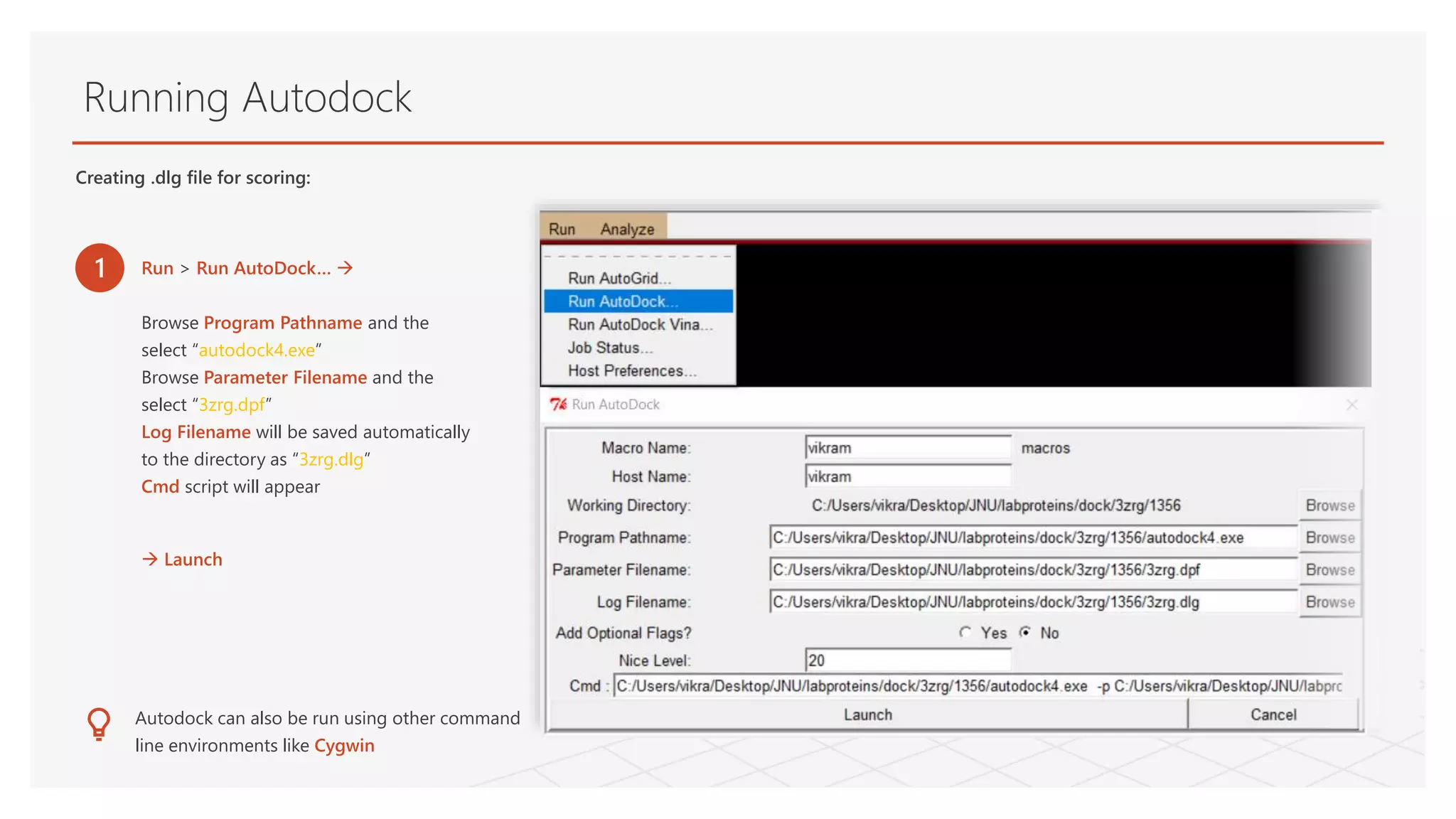
Running Autodock
Creating .dlgfile for scoring:
1 Run > Run AutoDock…
Browse Program Pathname and the
select “autodock4.exe”
Browse Parameter Filename and the
select “3zrg.dpf”
Log Filename will be saved automatically
to the directory as “3zrg.dlg”
Cmd script will appear
Launch
Autodock can also be run using other command
line environments like Cygwin
27.
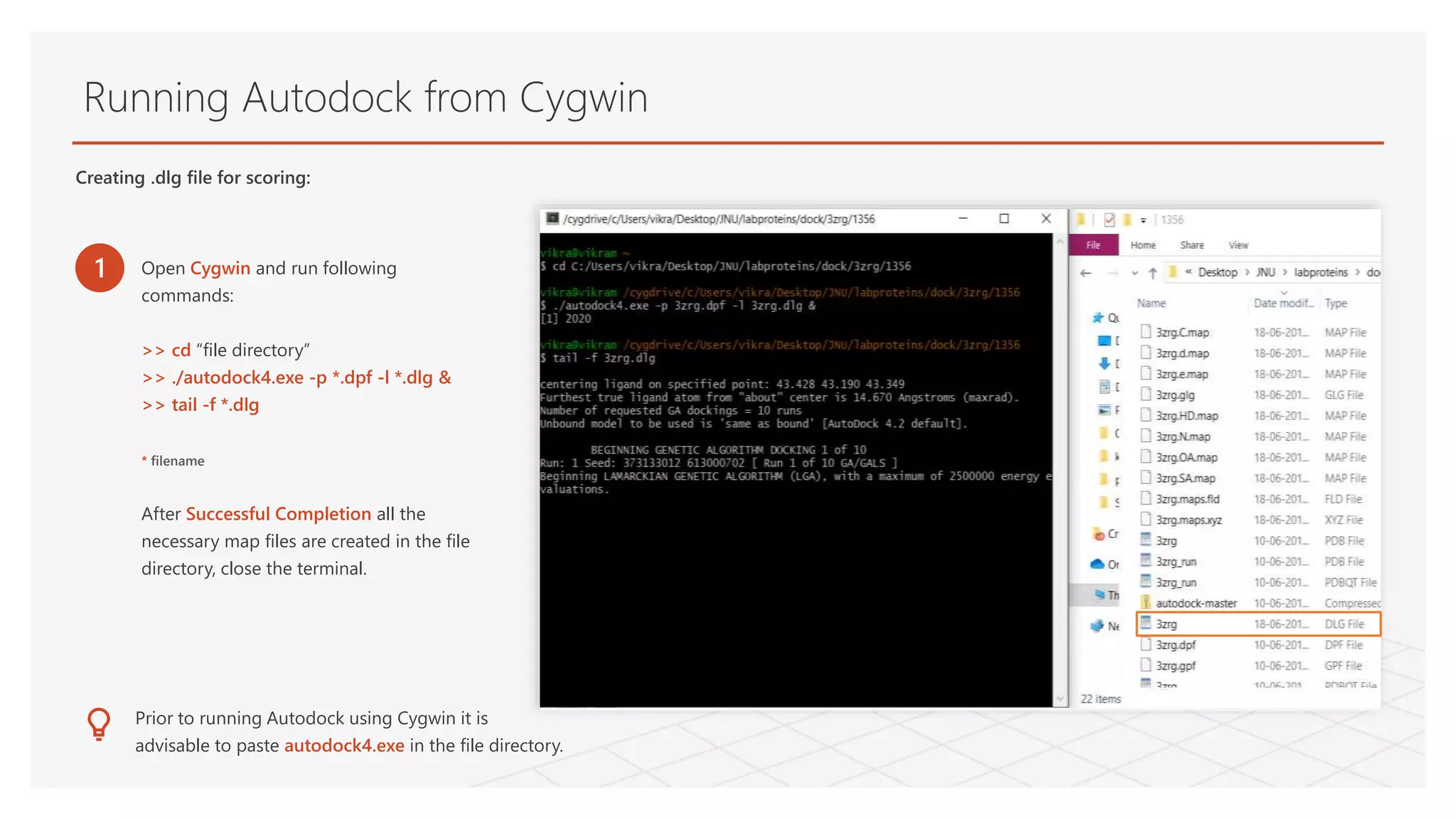
Running Autodock fromCygwin
1 Open Cygwin and run following
commands:
>> cd “file directory”
>> ./autodock4.exe -p *.dpf -l *.dlg &
>> tail -f *.dlg
* filename
After Successful Completion all the
necessary map files are created in the file
directory, close the terminal.
Prior to running Autodock using Cygwin it is
advisable to paste autodock4.exe in the file directory.
Creating .dlg file for scoring:
28.
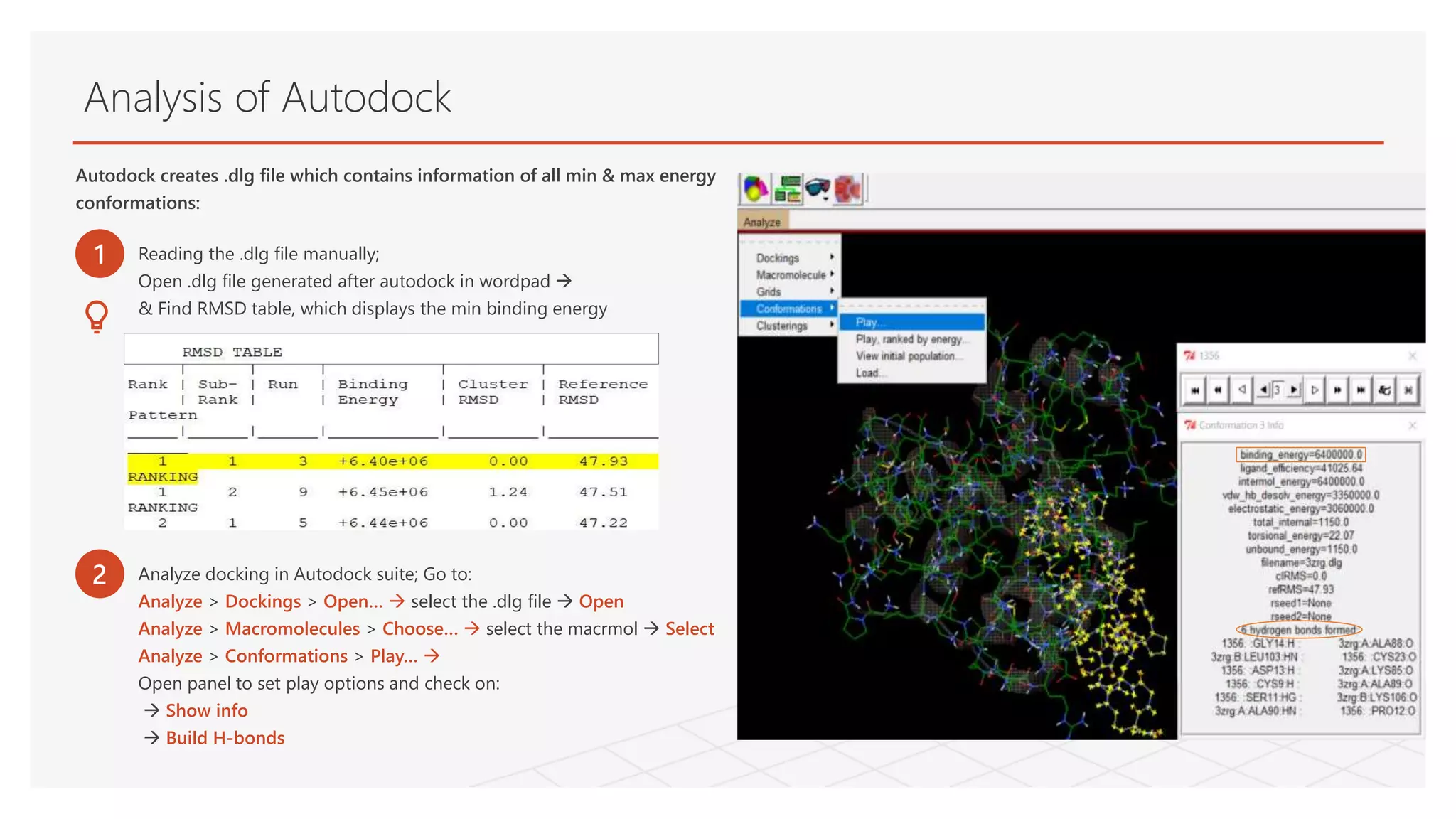
Analysis of Autodock
Autodockcreates .dlg file which contains information of all min & max energy
conformations:
1 Reading the .dlg file manually;
Open .dlg file generated after autodock in wordpad
& Find RMSD table, which displays the min binding energy
2 Analyze docking in Autodock suite; Go to:
Analyze > Dockings > Open… select the .dlg file Open
Analyze > Macromolecules > Choose… select the macrmol Select
Analyze > Conformations > Play…
Open panel to set play options and check on:
Show info
Build H-bonds


![Ligand
• Filtered FASTA sequence
• Structure prediction in QUARK ----> 3D structure (pdb)
• Structure Validation in PSVS
*Small peptide sequence ID: 0489
* this peptide is translated from non coding DNA ( junk DNA ) and modelled into 3D structure by
ab-inito approach [Ref: Dr. Pawan Dhar, Synthetic Biology Lab, School of Biotechnology, JNU]](https://image.slidesharecdn.com/moleculardockingautodock-200109085702/75/Molecular-Docking-Using-Autodock-Tools-6-2048.jpg)