Downloaded 45 times





![Here as an example we are drawing the structure of DB75 [2,5-bis(4-
amidinophenyl)furan], also known as furamidine, is an analog of pentamidine.
“Optimize Geometry” option which can be located from “Extensions” tab as
shown in Figure, allows the user to optimize the structure of the molecule to
its stable conformation.](https://image.slidesharecdn.com/7energyminimization-191130121614/85/Energy-minimization-7-320.jpg)

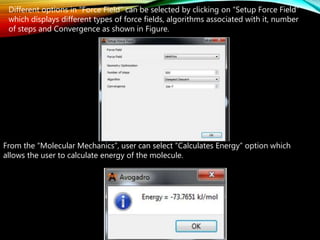
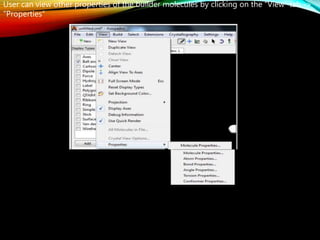

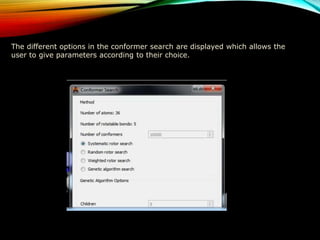
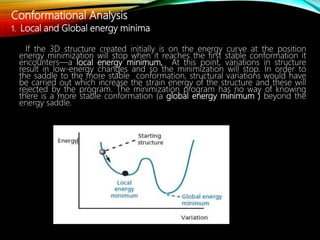
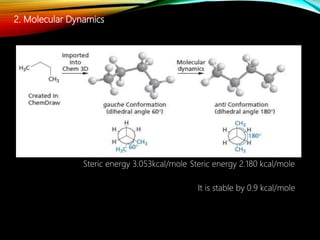



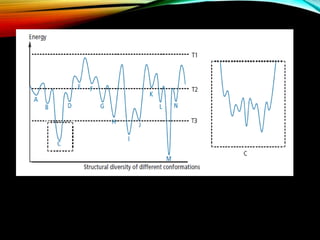

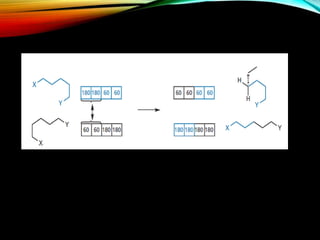


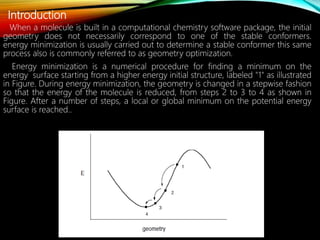
The document discusses methods and procedures for energy minimization in computational chemistry, focusing on geometry optimization to achieve stable molecular conformers. Various techniques including the Newton-Raphson, steepest descent, and conjugate gradient methods are outlined, along with practical applications and tools available in software for molecular mechanics and conformational analysis. It also covers concepts like local and global energy minima, Monte Carlo methods, and genetic algorithms for conformational exploration.






































![MINIMUM_ENERGY_CONFORMATIONS[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/minimumenergyconformations1-230901171431-bceffb16-thumbnail.jpg?width=640&height=640&fit=bounds)










![TITANIUM CHLORIDE [PHARMACEUTICAL REAGENT]](https://cdn.slidesharecdn.com/ss_thumbnails/66-191220064235-thumbnail.jpg?width=640&height=640&fit=bounds)
![OXIDATION [PHARMACEUTICAL PROCESS CHEMISTRY]](https://cdn.slidesharecdn.com/ss_thumbnails/65-191220062805-thumbnail.jpg?width=640&height=640&fit=bounds)
![Dicyclohexylcarbodiimide [DCC]](https://cdn.slidesharecdn.com/ss_thumbnails/61-191219150428-thumbnail.jpg?width=640&height=640&fit=bounds)
![Homogeneous catalysis [ MPHARM, MSC, BPHARM, BSC]](https://cdn.slidesharecdn.com/ss_thumbnails/62-191219155346-thumbnail.jpg?width=640&height=640&fit=bounds)
![OLIGONUCLEOTIDE THERAPY [ TECHNIQUES, APPLICATIONS]](https://cdn.slidesharecdn.com/ss_thumbnails/66-191220063512-thumbnail.jpg?width=640&height=640&fit=bounds)
![PHASE TRANSFER CATALYSIS [PTC]](https://cdn.slidesharecdn.com/ss_thumbnails/64-191219161253-thumbnail.jpg?width=640&height=640&fit=bounds)
![MUTUAL PRODRUG [PHARMACEUTICALS]](https://cdn.slidesharecdn.com/ss_thumbnails/67-191220064837-thumbnail.jpg?width=640&height=640&fit=bounds)


![LIQUID CHROMATOGRAPHY- MASS SPECTROSCOPY[LC-MS]](https://cdn.slidesharecdn.com/ss_thumbnails/63-191219160156-thumbnail.jpg?width=640&height=640&fit=bounds)




![problems associated with skin[ as per Pharmaceutics]](https://cdn.slidesharecdn.com/ss_thumbnails/2-200204102918-thumbnail.jpg?width=640&height=640&fit=bounds)
![cosmetics and cosmeticals [department of pharmaceutics]](https://cdn.slidesharecdn.com/ss_thumbnails/1-200204101110-thumbnail.jpg?width=640&height=640&fit=bounds)






















