Downloaded 221 times





![Classification of Amyloidosis:
Systemic (Generalized) Amyloidosis:
primary amyloidosis:
o Immunocyte dyscrasias with amyloidosis:
as in Multiple myeloma and other monoclonal
B-cell proliferations [ AL protein ].
secondary amyloidosis:
o Chronic inflammatory conditions such as T.B,
bronchiectasis, and osteomyelitis [ AA protein ].
o Hemodialysis-associated amyloidosis in
Chronic renal failure[ β2-microglobulin( Aβ2m )].](https://image.slidesharecdn.com/immunopathology-6-130219045949-phpapp02/85/Immunopathology-6-5-320.jpg)
![ Hereditary amyloidosis:
o Familial Mediterranean fever [ AA protein ].
o Familial amyloidotic neuropathies [ ( ATTR )
Transthyretin amyloid ].
Systemic senile amyloidosis:
[ (ATTR) Transthyretin amyloid ].
Localized Amyloidosis:
Senile cerebral Alzheimer disease [ β-amyloid
protein (Aβ) ].
Medullary carcinoma of thyroid [ Calcitonin ].](https://image.slidesharecdn.com/immunopathology-6-130219045949-phpapp02/85/Immunopathology-6-6-320.jpg)



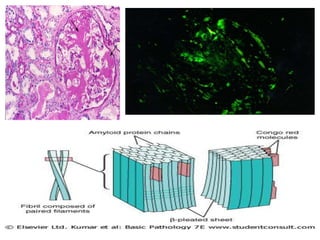


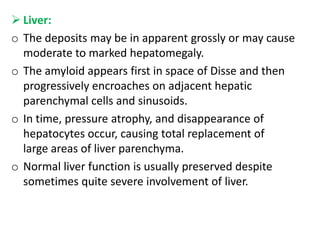







Amyloidosis is a group of diseases caused by abnormal protein deposits in tissues. There are different types of amyloid proteins that can deposit, including immunoglobulin light chains (AL), serum amyloid A (AA), and beta-amyloid. The deposits disrupt organ function over time and can affect the kidneys, heart, liver, spleen, and tongue. Symptoms vary by the organs involved but may include kidney failure, heart failure, digestive issues, and enlarged tongue. The prognosis depends on the type and extent of amyloidosis. Treatment focuses on managing the underlying condition that causes protein deposition or targeting the amyloid deposits directly.








































































![Prac excises 3[1].5](https://cdn.slidesharecdn.com/ss_thumbnails/pracexcises31-150331131154-conversion-gate01-thumbnail.jpg?width=640&height=640&fit=bounds)













