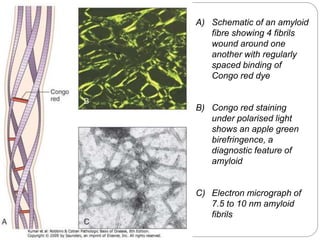
Amyloidosis is a condition caused by abnormal protein deposits forming fibrils that damage tissues. These fibrils result from improperly folded proteins aggregating. There are different types depending on the precursor protein, including AL amyloidosis associated with plasma cell disorders and AA amyloidosis linked to chronic inflammation. Amyloid fibrils have a characteristic beta-pleated sheet structure and stain apple-green under polarized light when bound to Congo red dye. The deposits can affect many organs and cause tissue destruction and organ dysfunction. The kidneys, liver, spleen, heart and nervous system are commonly involved.