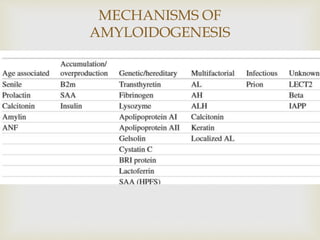
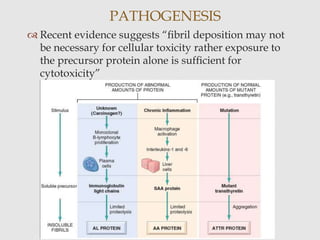
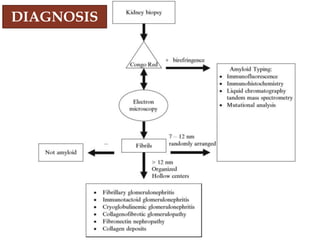
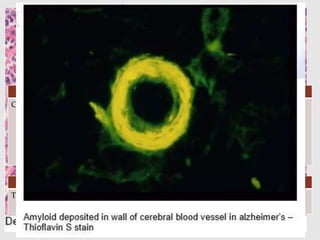
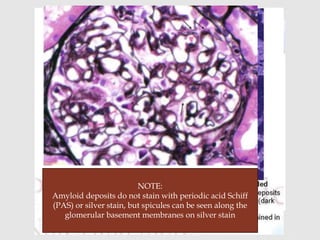
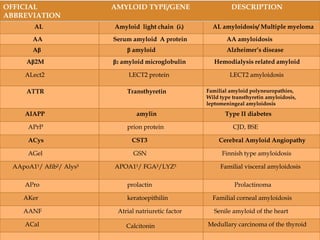
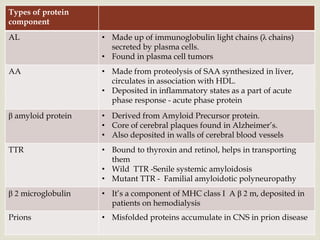
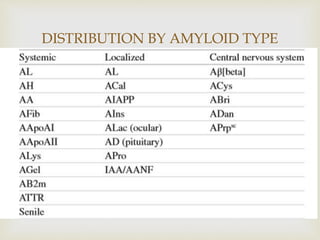
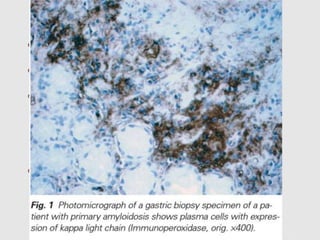
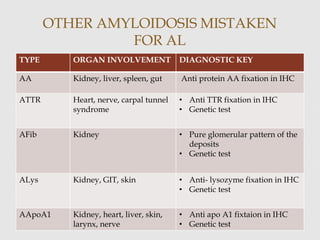
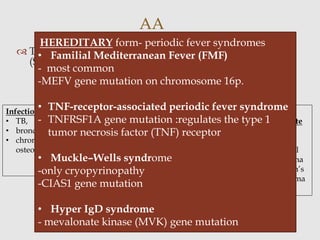
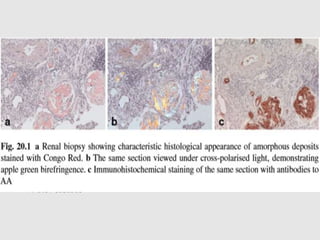
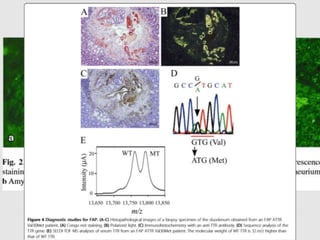
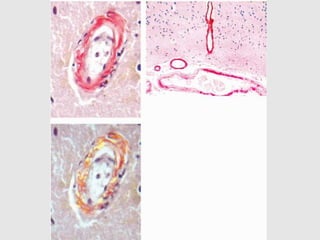
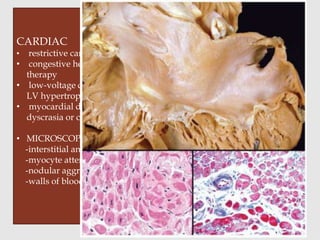
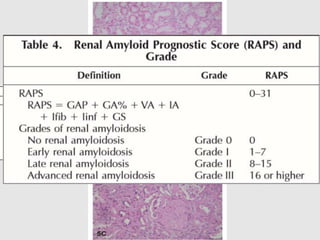
Dr. Sadaf Khan discusses amyloidosis, a disorder caused by the deposition of abnormal protein fibrils in tissues and organs. Amyloidosis can be localized to a single organ or systemic. The type of amyloid protein deposited determines the classification, with the most common types being AL amyloidosis associated with plasma cell dyscrasias and AA amyloidosis associated with inflammatory conditions. Diagnosis involves staining biopsy samples with dyes like Congo red and examining under polarized light. Treatment depends on the type and organ involvement but may include targeting the underlying condition, chemotherapy, dialysis, or organ transplantation.