Downloaded 183 times





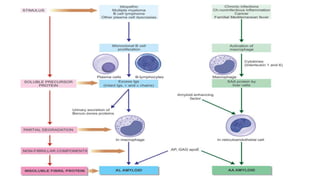
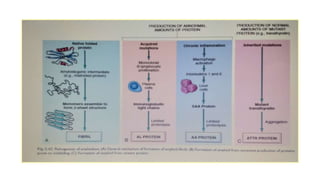
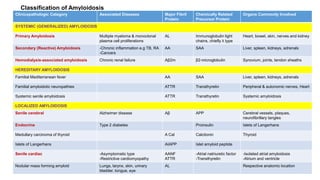


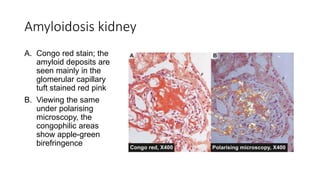
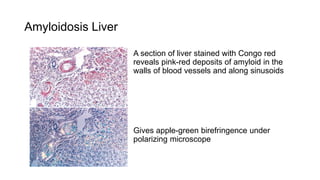
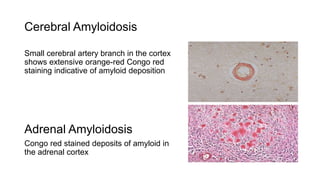
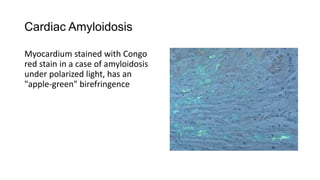




This document discusses amyloidosis, including: - Amyloidosis occurs when proteins aggregate into insoluble fibrils called amyloid that deposit abnormally in tissues. Over 30 proteins can form amyloid fibrils. - Deposition can result from excessive protein production, mutations causing improper folding, or defective degradation of extracellular proteins. Deposits damage tissues. - The main types are AL amyloidosis from immunoglobulin light chains, AA amyloidosis from serum amyloid A, and ATTR amyloidosis from transthyretin. - Amyloidosis can be systemic, affecting multiple organs, or localized to single organs. Common sites for biopsy include kidney, rectum, abdominal fat. Congo red staining demonstrates amyloid's pink























































































