Downloaded 123 times

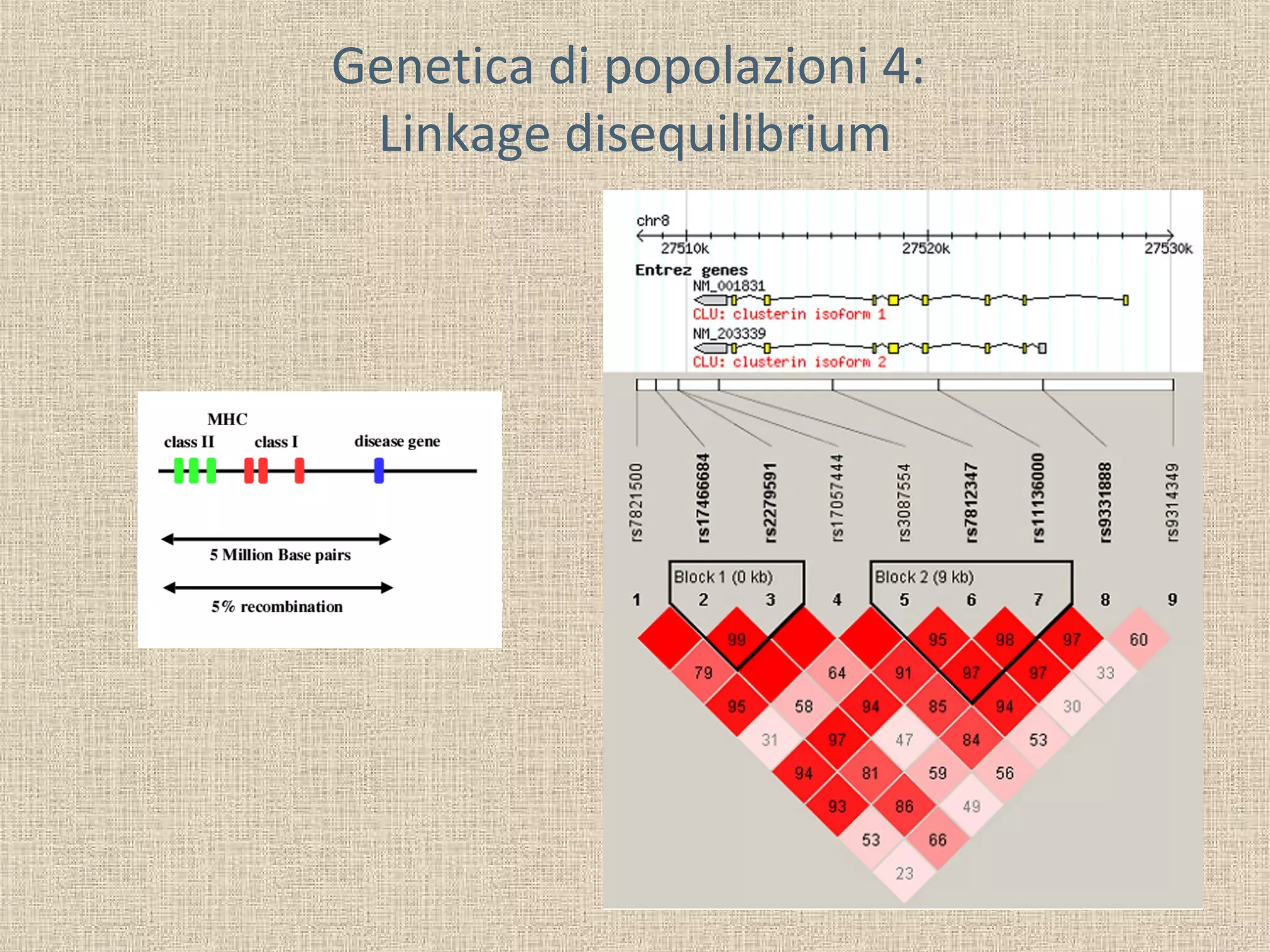


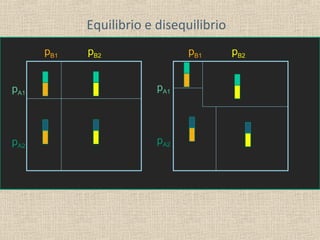
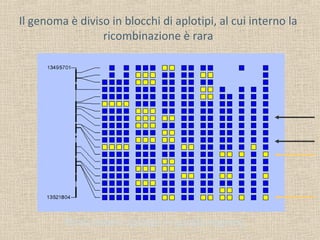
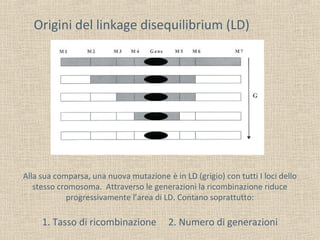

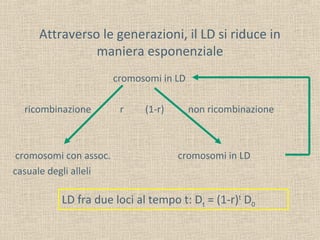
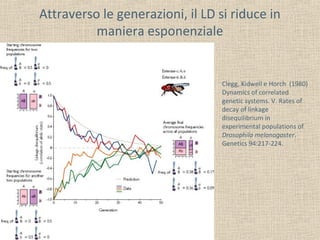
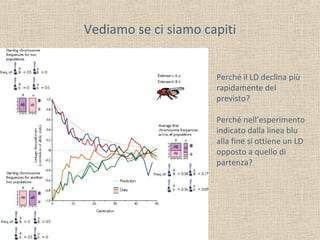
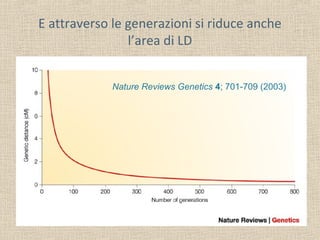








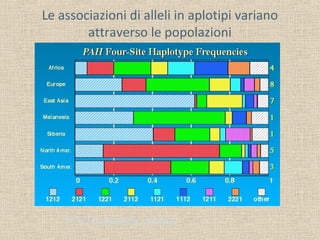
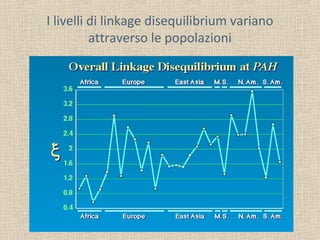
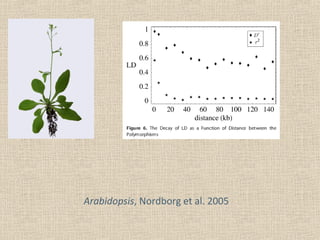
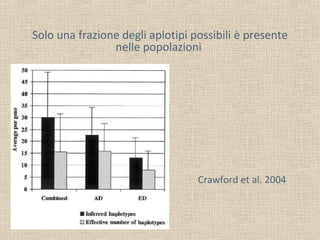
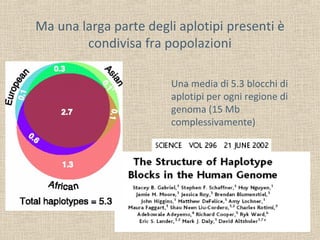
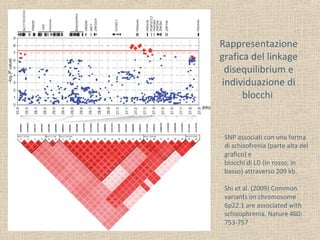



Il documento tratta della genetica di popolazioni, focalizzandosi sul linkage disequilibrium (LD) e i suoi fondamenti, come l'equilibrio di Hardy-Weinberg, la mutazione e la ricombinazione. Descrive come il LD e la sua riduzione nel tempo dipendano dai tassi di ricombinazione e dal numero di generazioni, sottolineando l'importanza dei aplotipi. Inoltre, evidenzia che, mentre l'equilibrio di Hardy-Weinberg può essere raggiunto rapidamente, il linkage equilibrium richiede più tempo e può essere analizzato tramite diverse statistiche.













































