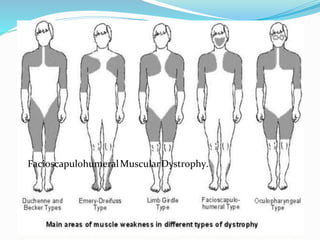
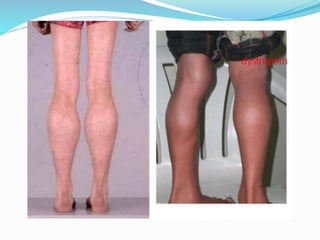
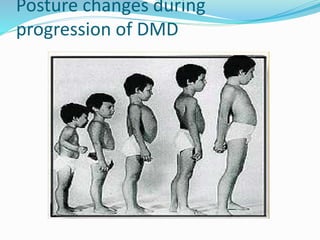
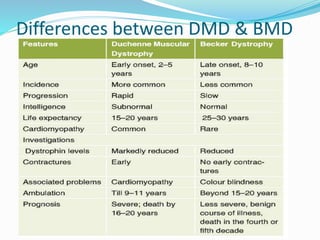
This document discusses Duchenne muscular dystrophy (DMD), the most common form of muscular dystrophy. DMD is an X-linked recessive genetic disorder caused by mutations in the dystrophin gene. This results in progressive muscle degeneration and weakness. The document outlines the characteristic features and stages of DMD as well as methods of genetic testing, management, and potential exon skipping treatments that are being investigated.