Downloaded 2,346 times














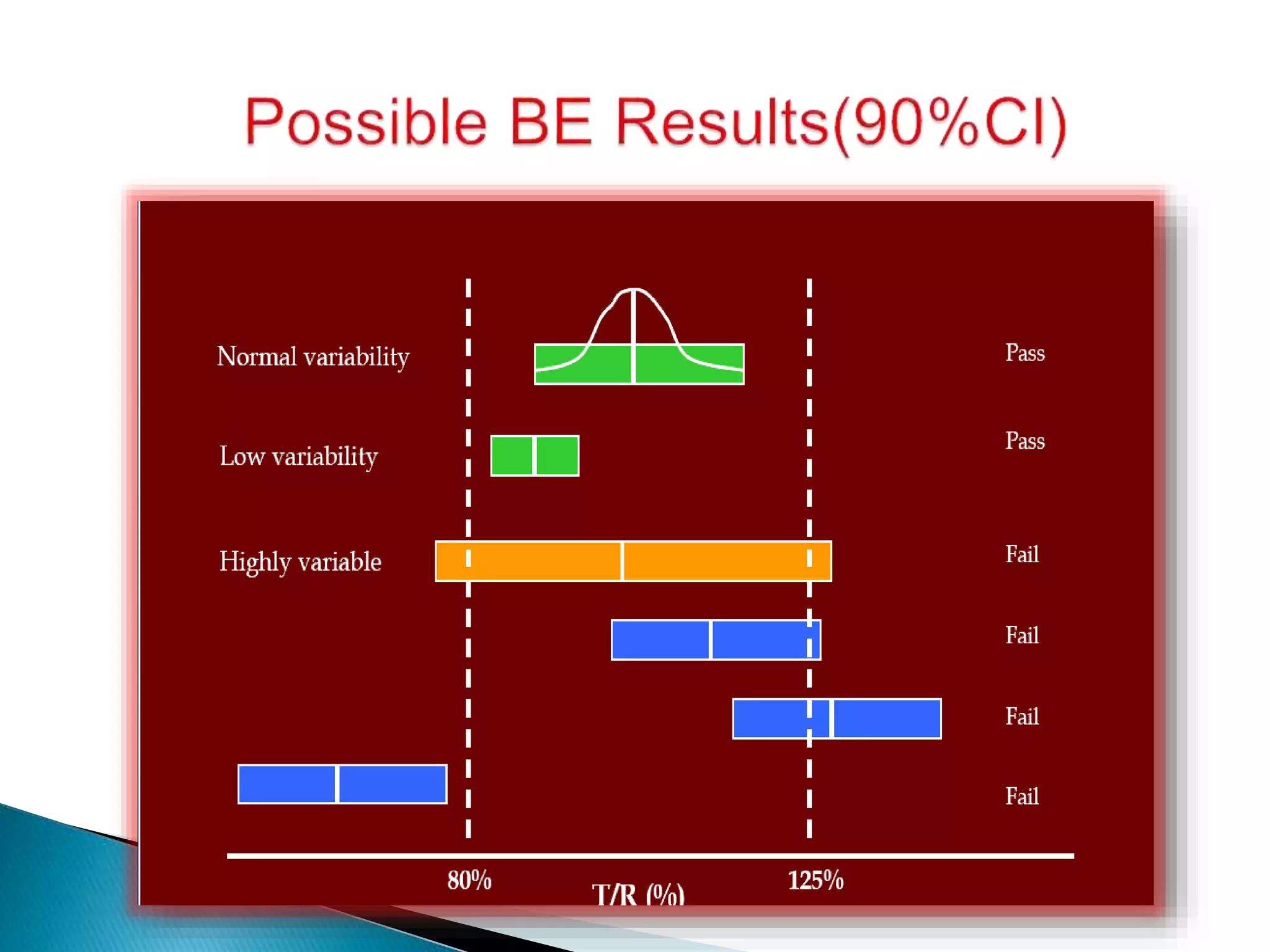
![[ Criterion ] BE Limit
Average BE: (T - R)2 qA
2
(T - R)2 + D
2 + (WT
2 - WR
2)
Individual BE: ------------------------------------------- qI
WR
2
(T - R)2 + (TT
2 - TR
2)
Population BE: ----------------------------------- qP
TR
2](https://image.slidesharecdn.com/babe-150203065431-conversion-gate01/75/Bioavailability-and-Bioequivalence-Studies-BABE-Concept-of-Biowaivers-17-2048.jpg)



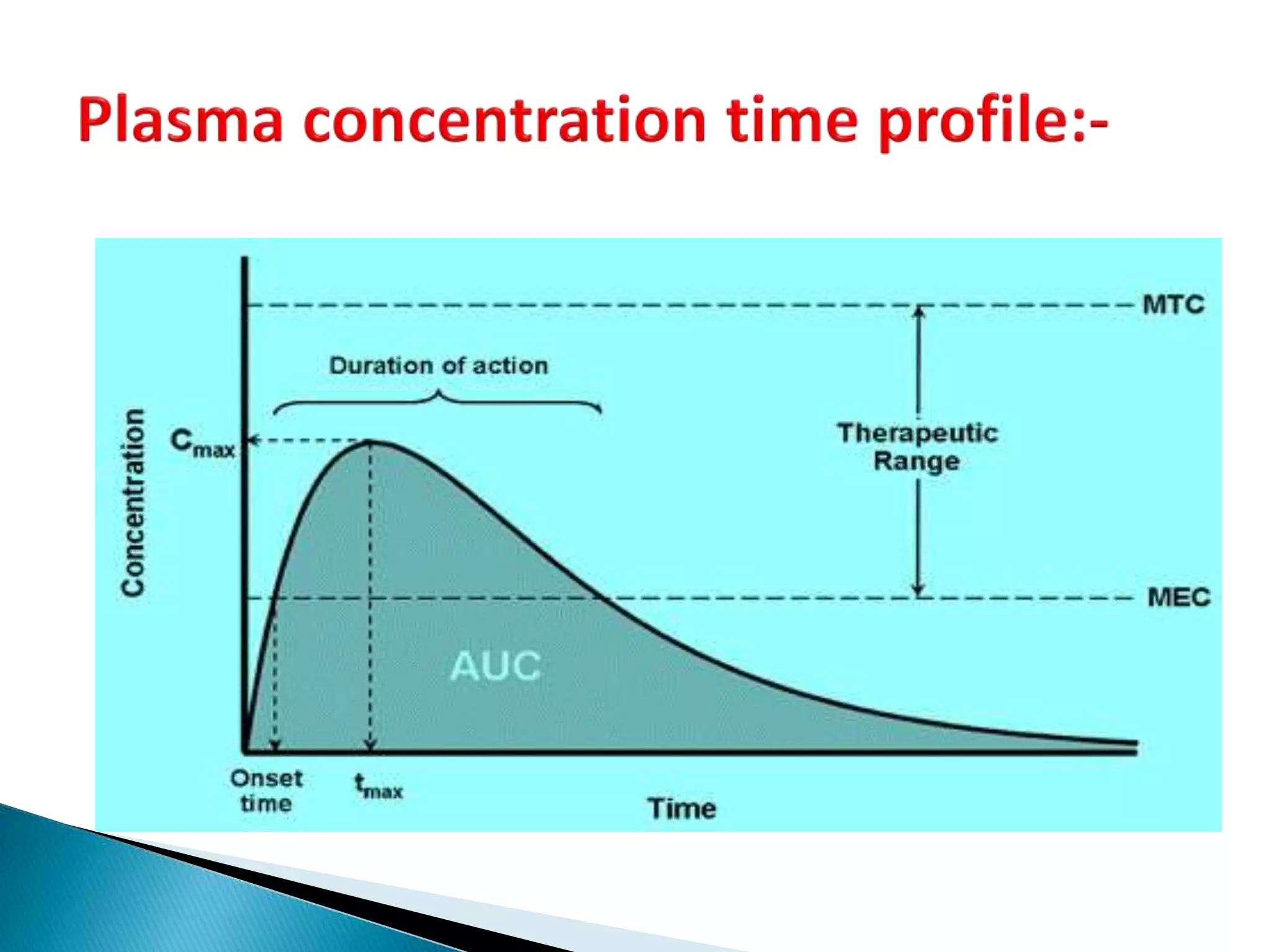


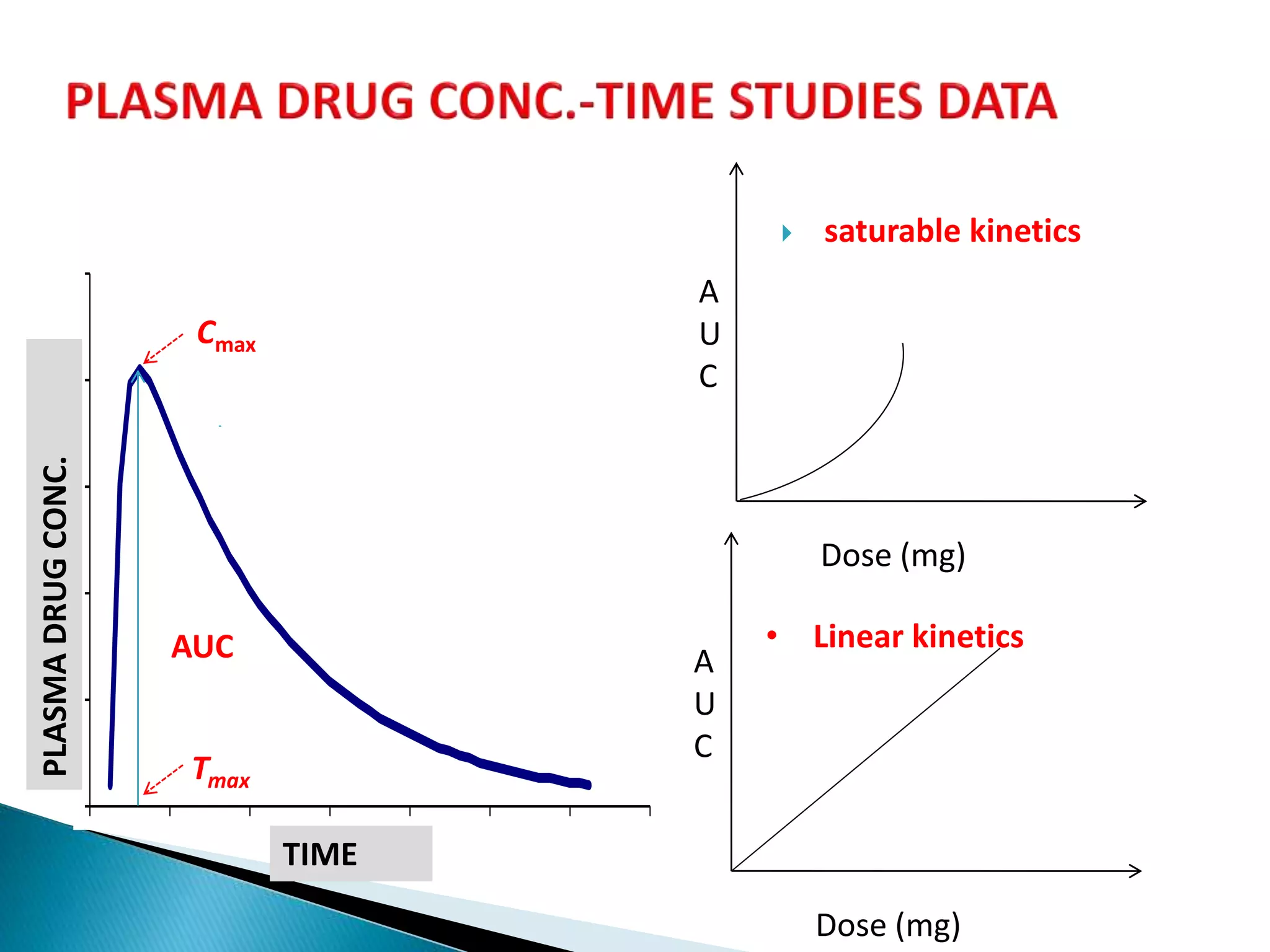
![Cmax
PLASMADRUGCONC.
TIME
Tmax
AUC
oral
iv
iv
oral
Dose
Dose
AUC
AUC
F
stdtest
teststd
std
test
D
D
AUC
AUC
F
][
][
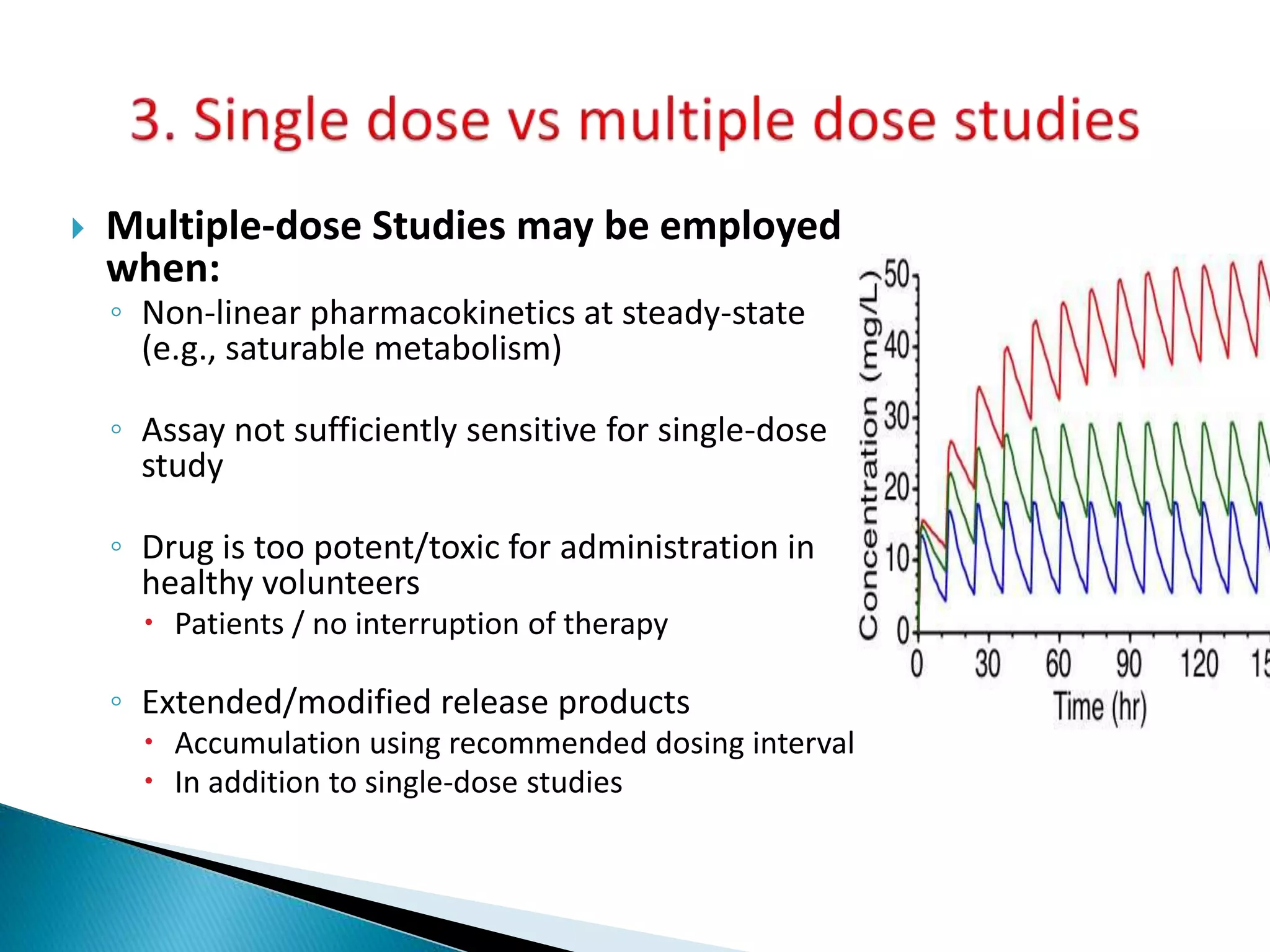
Single dose studies
Multiple dose studies
...](https://image.slidesharecdn.com/babe-150203065431-conversion-gate01/75/Bioavailability-and-Bioequivalence-Studies-BABE-Concept-of-Biowaivers-25-2048.jpg)
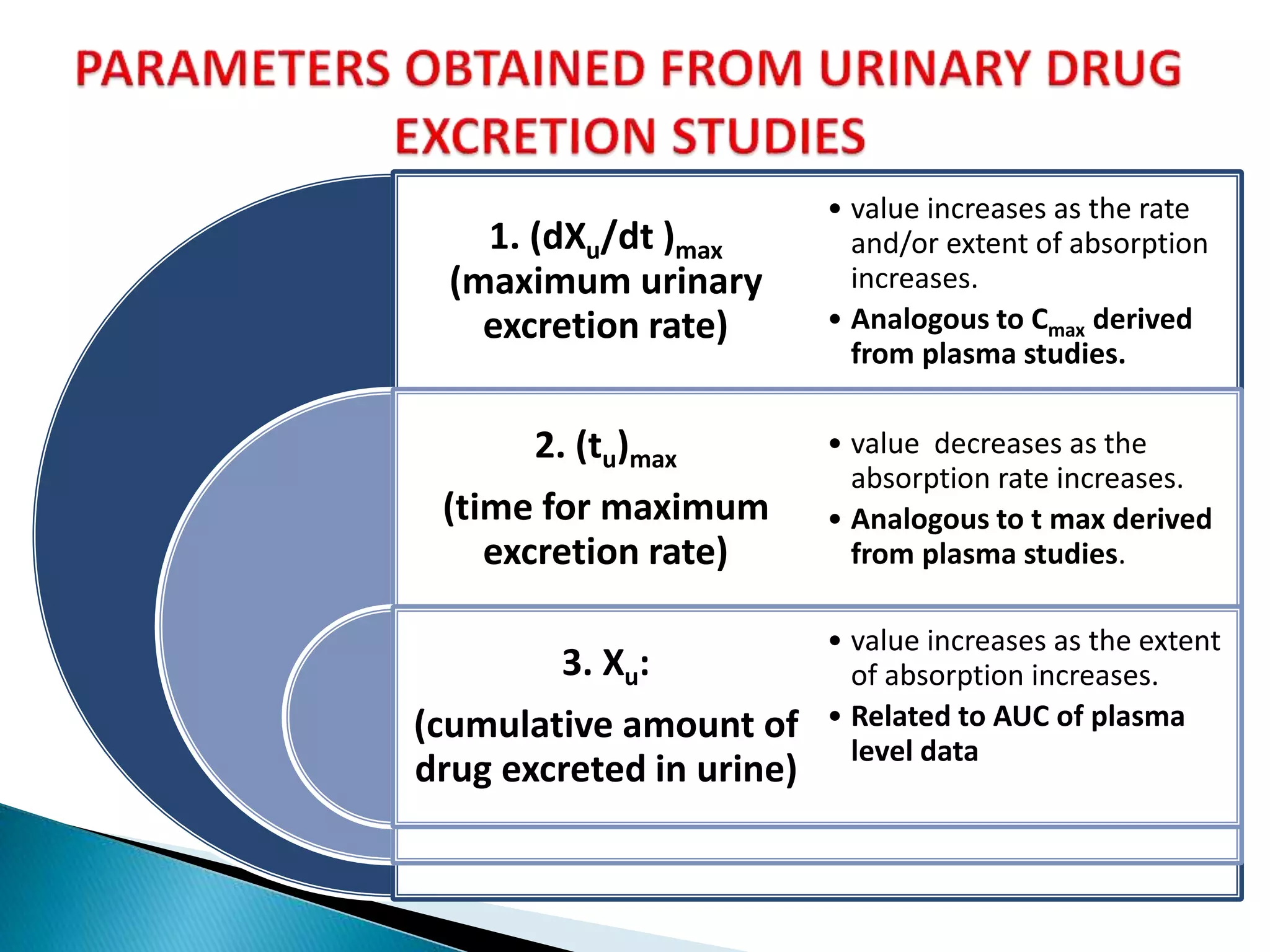
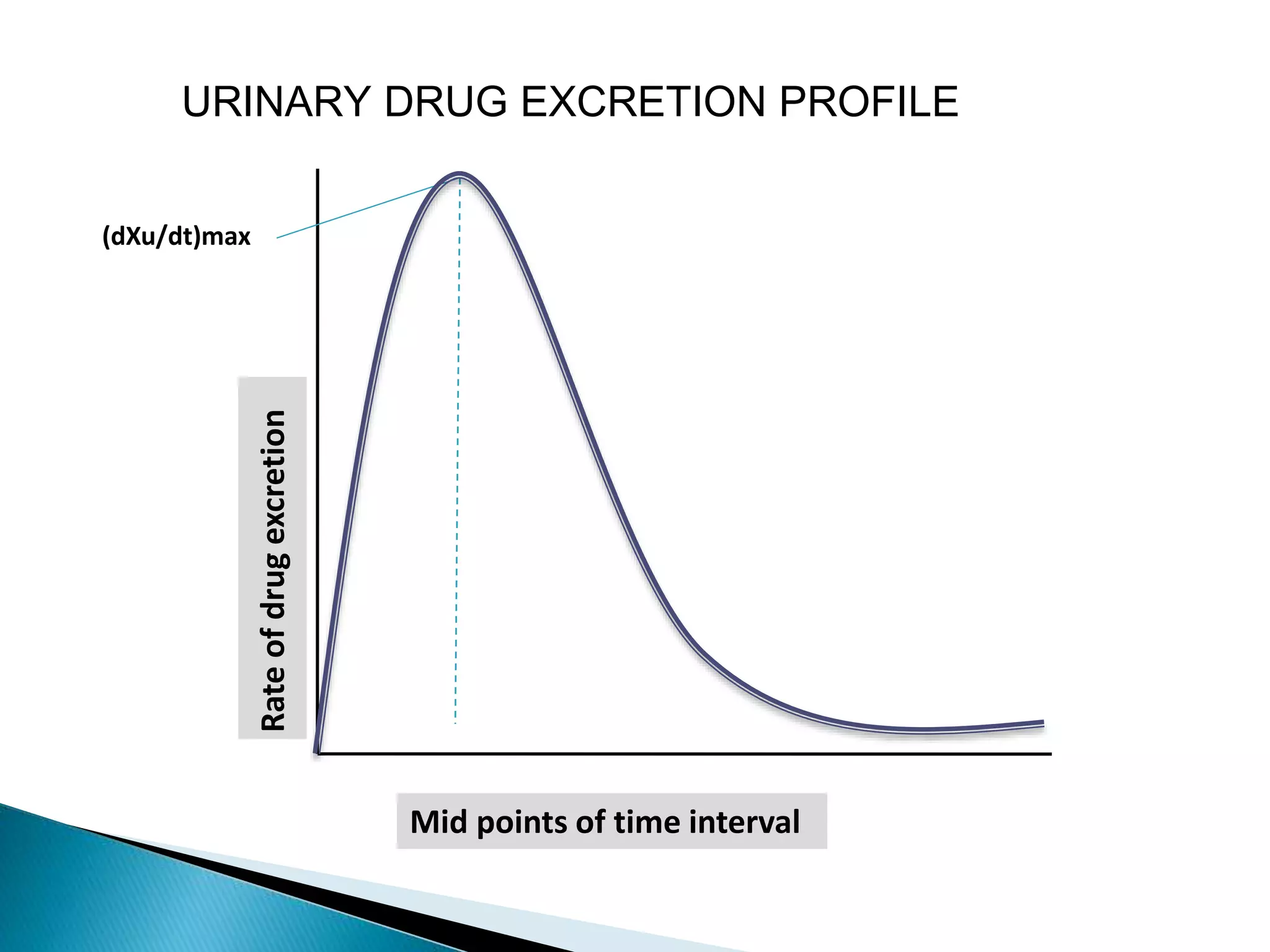
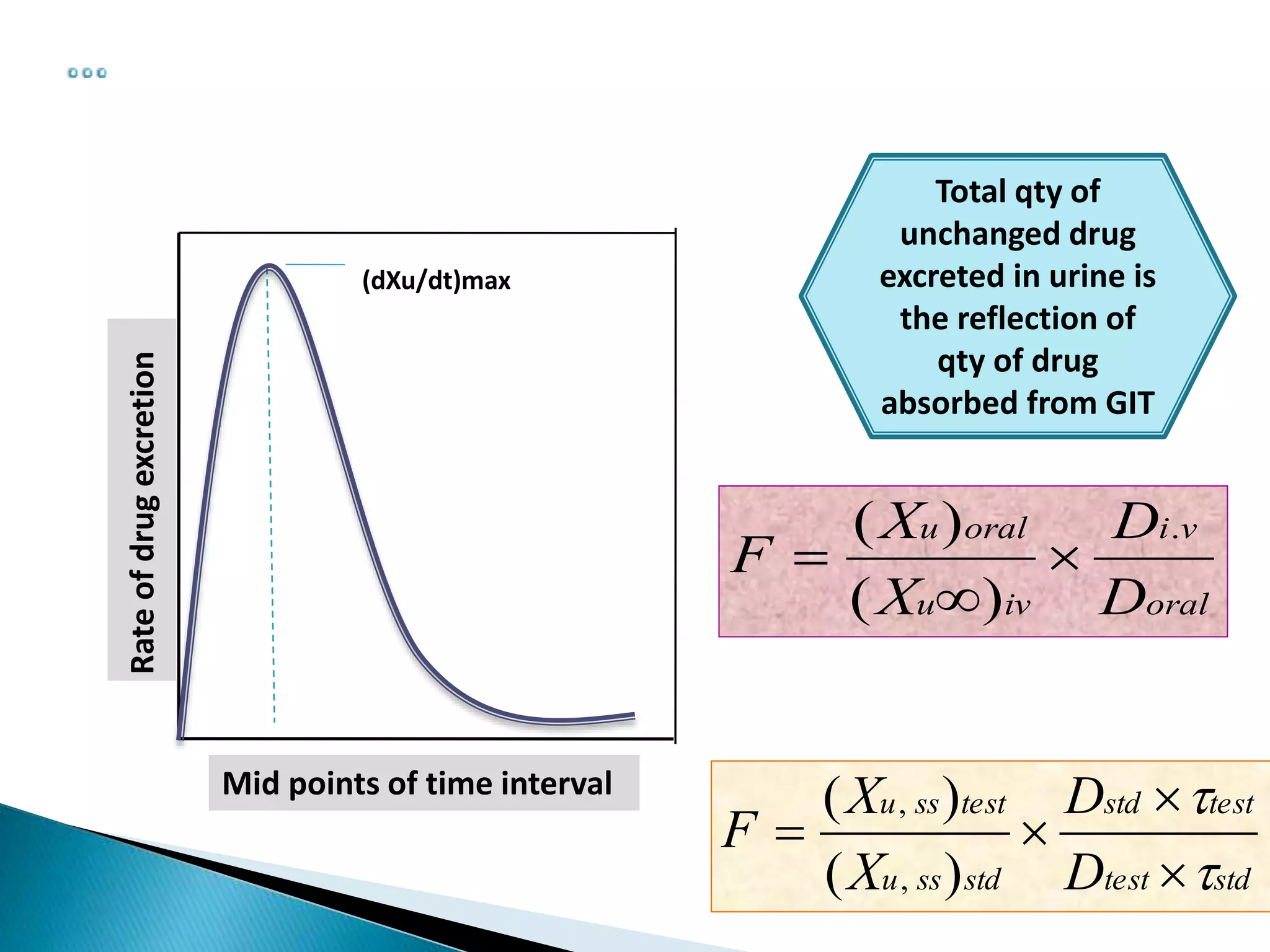















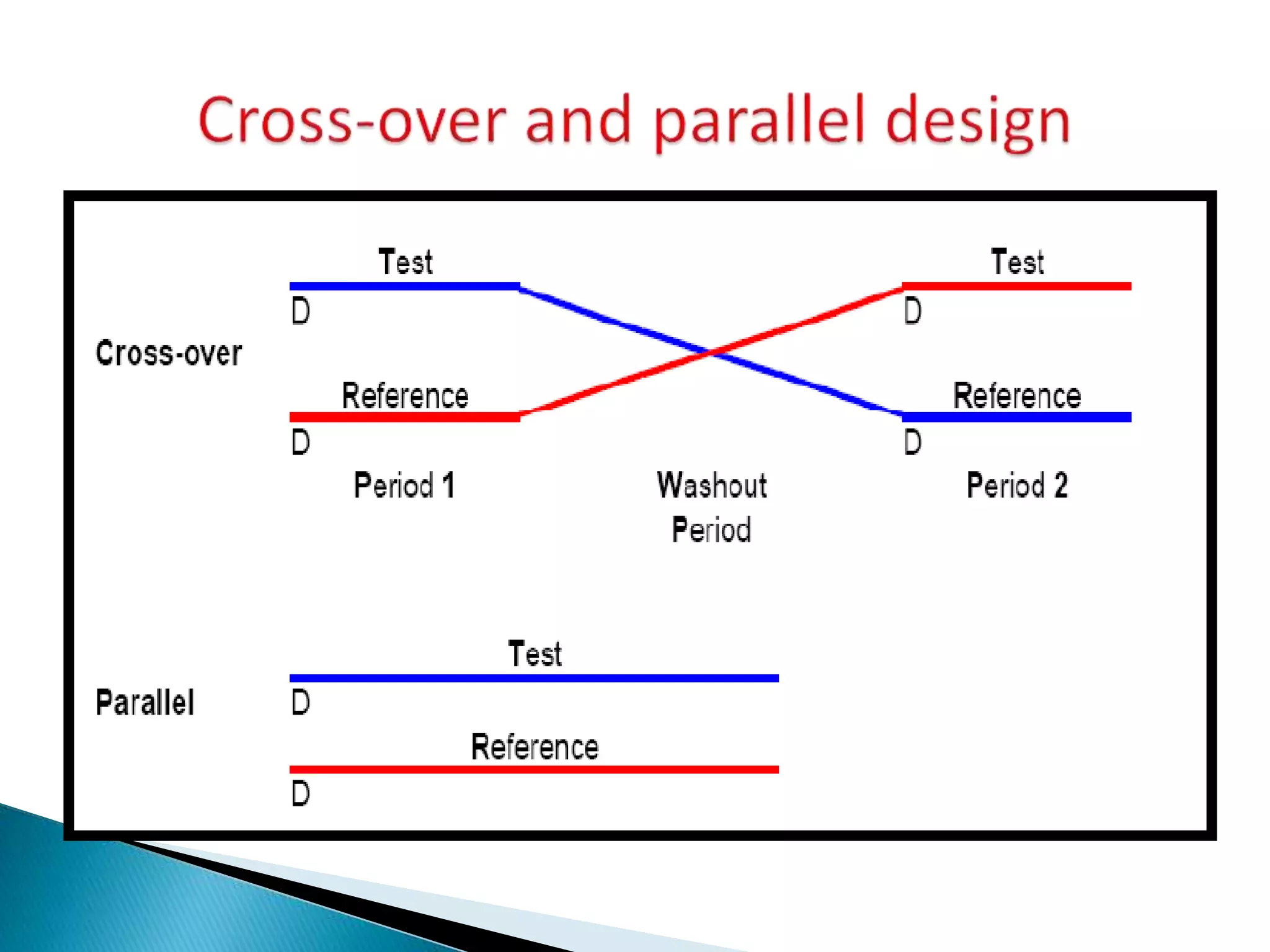
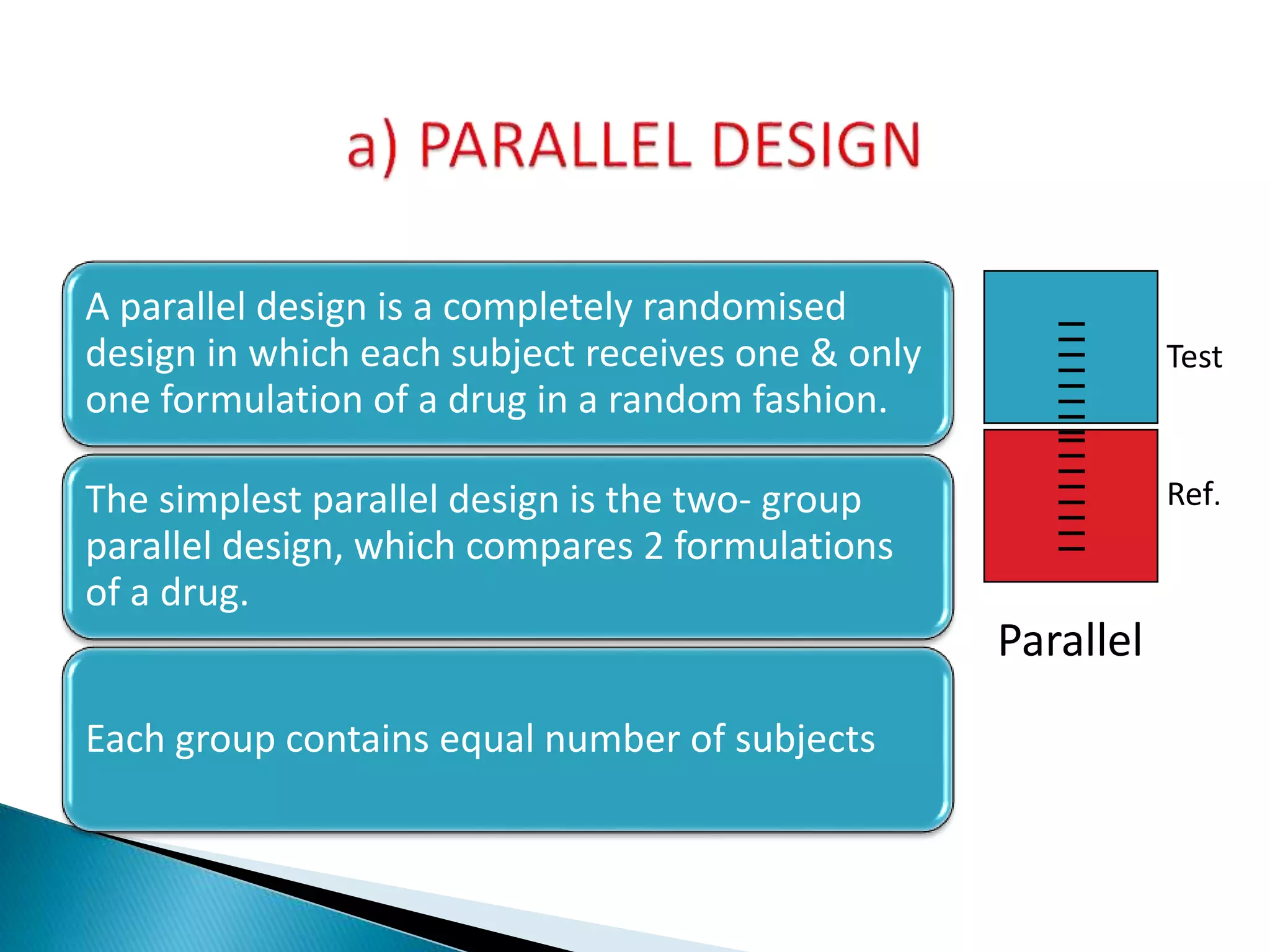

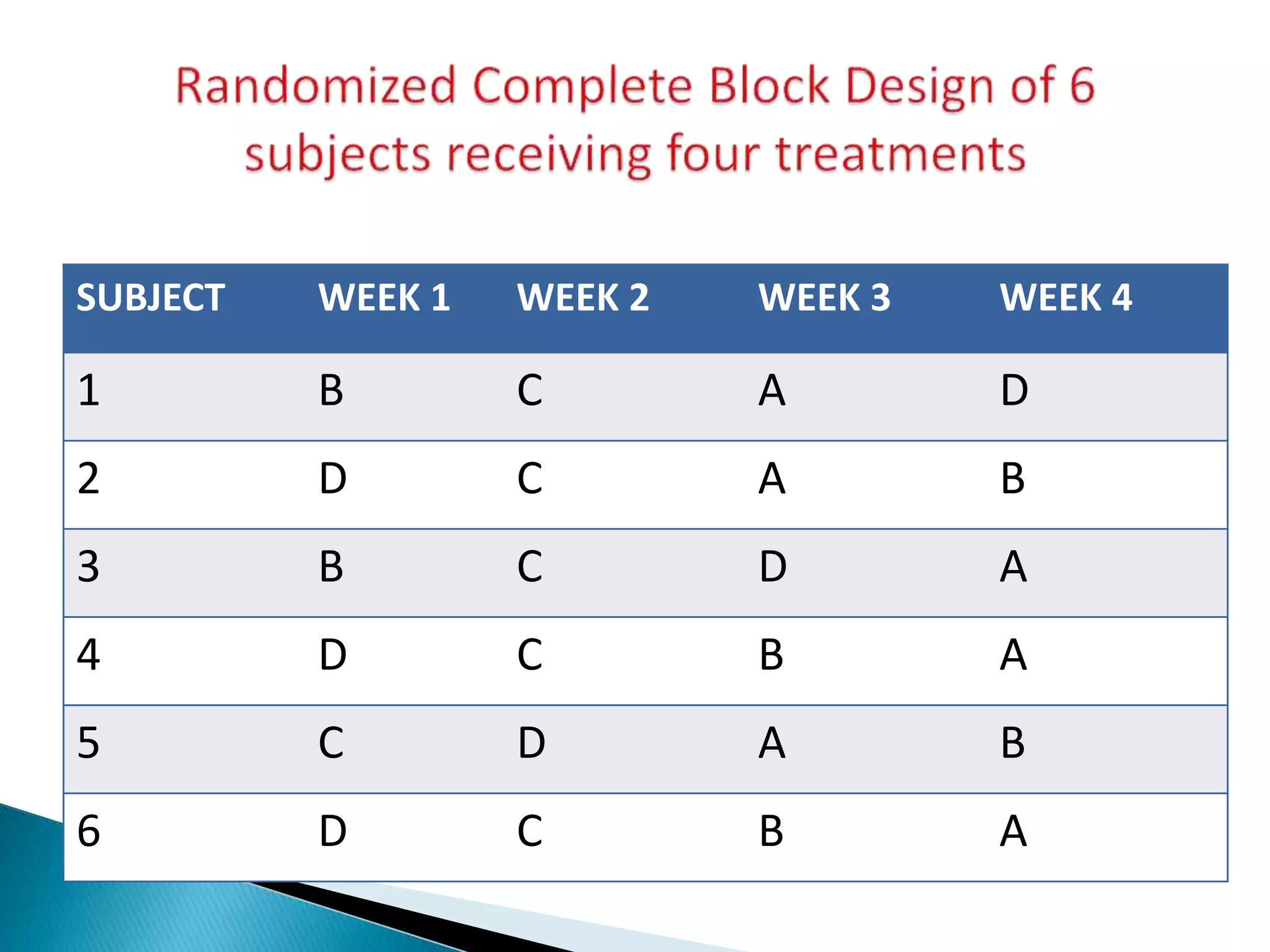
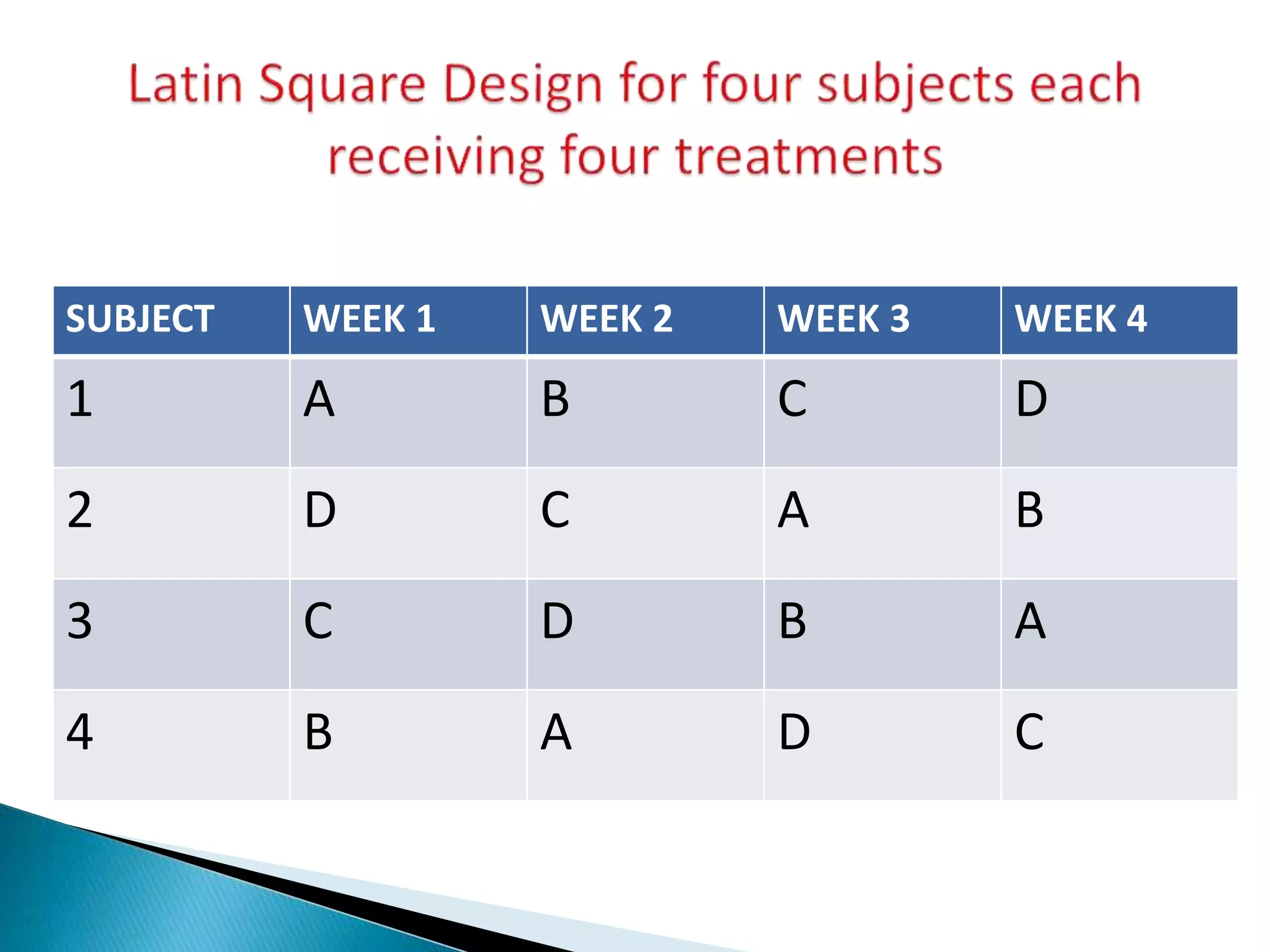
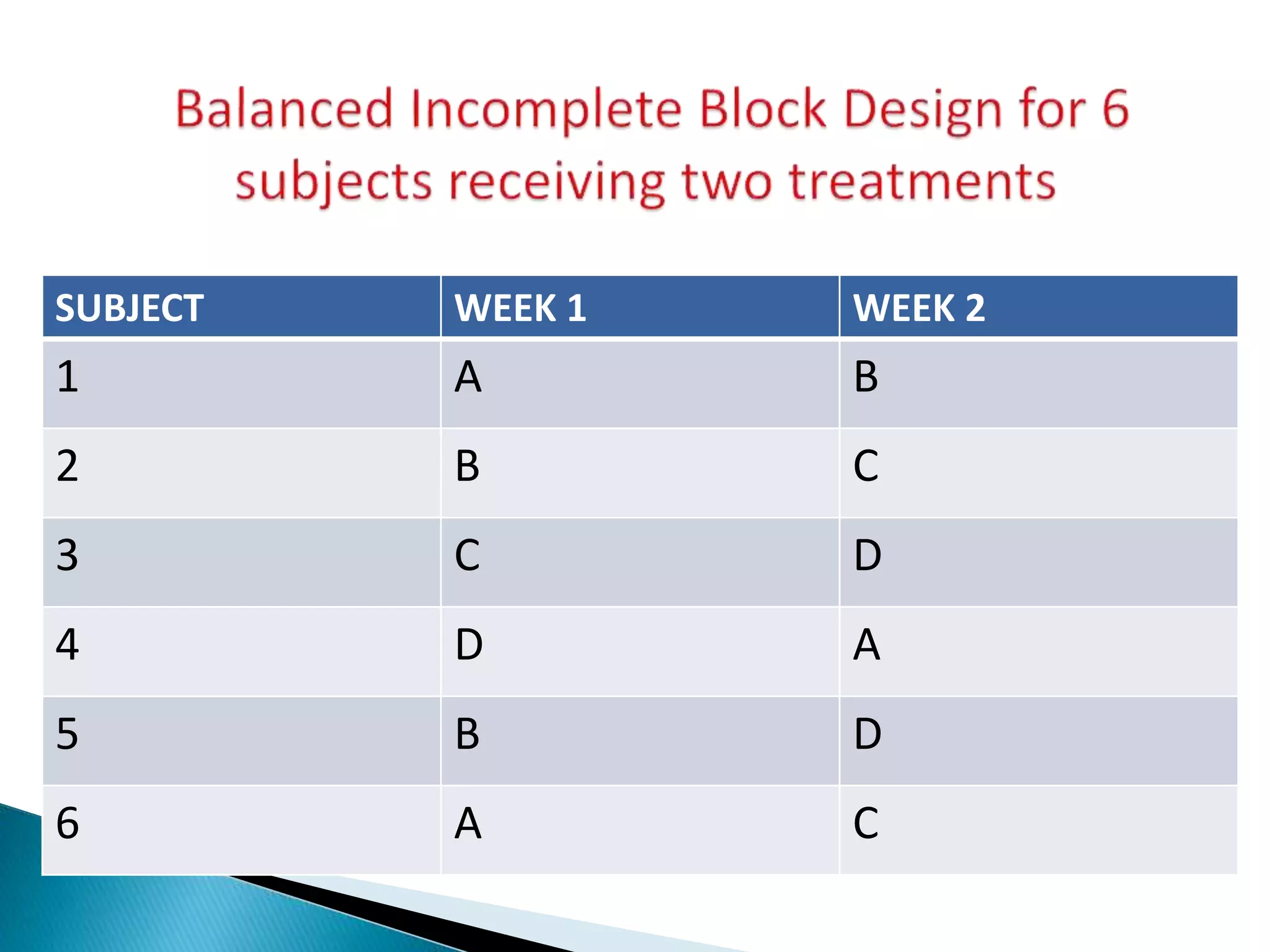
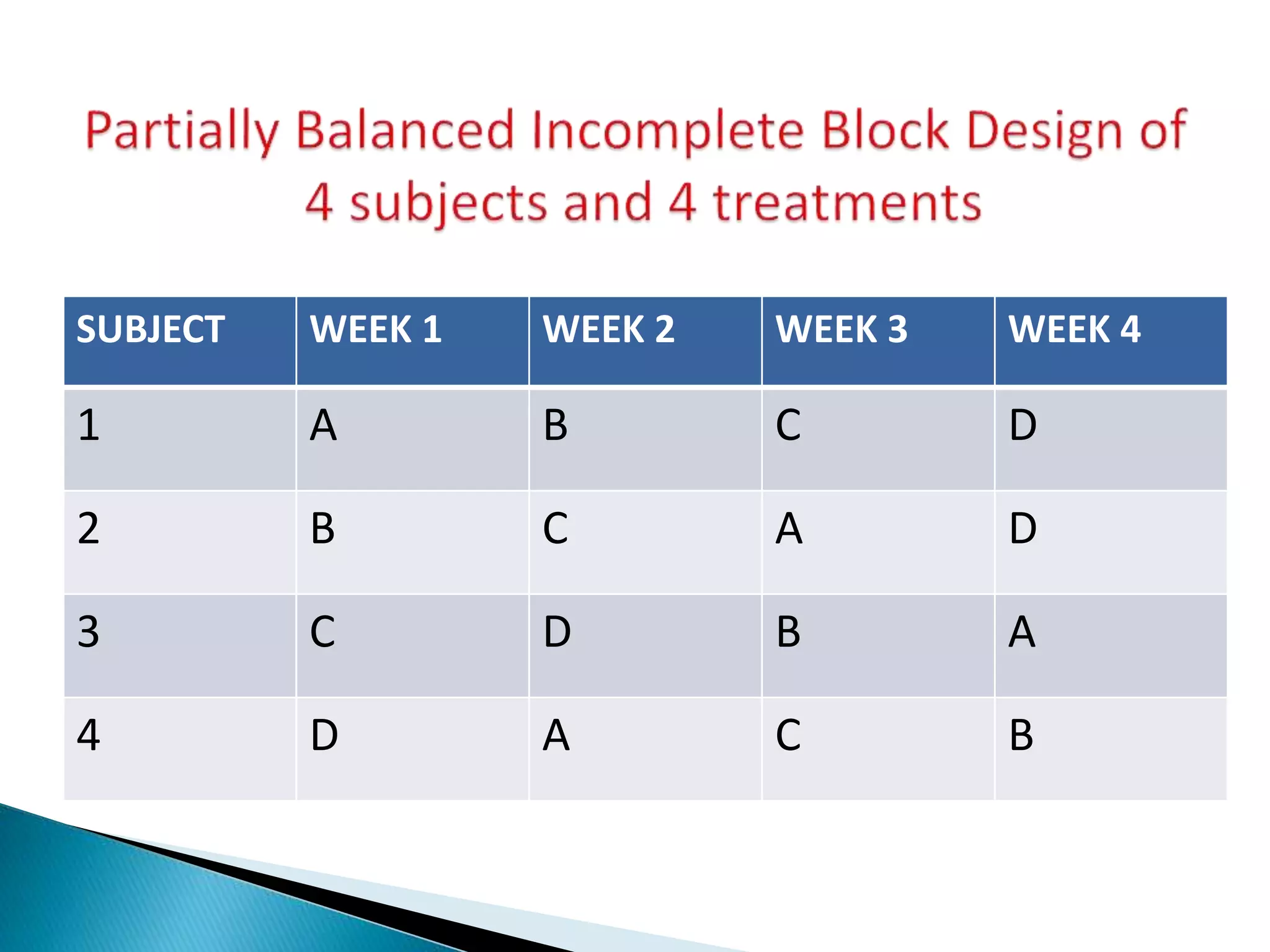


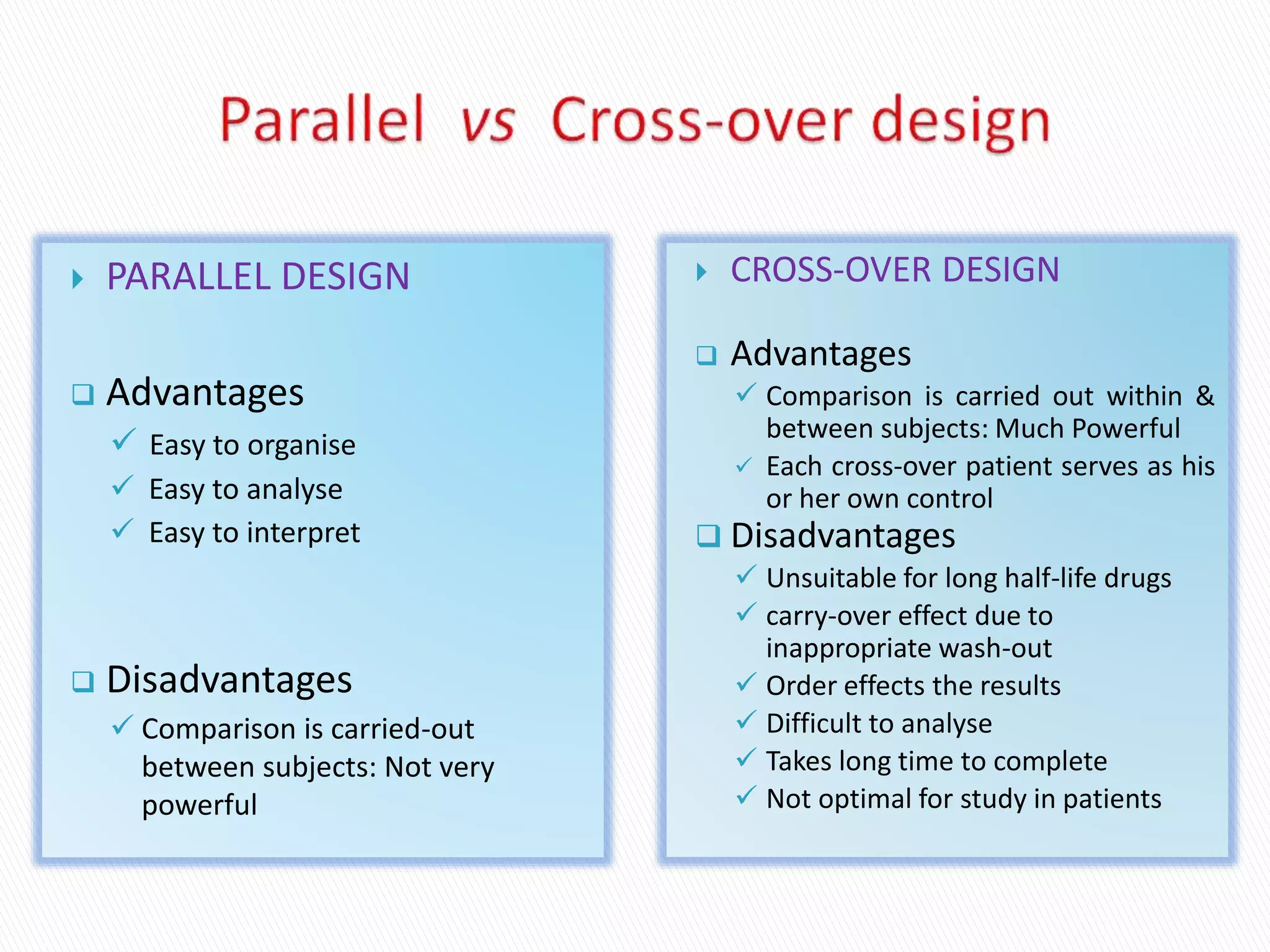
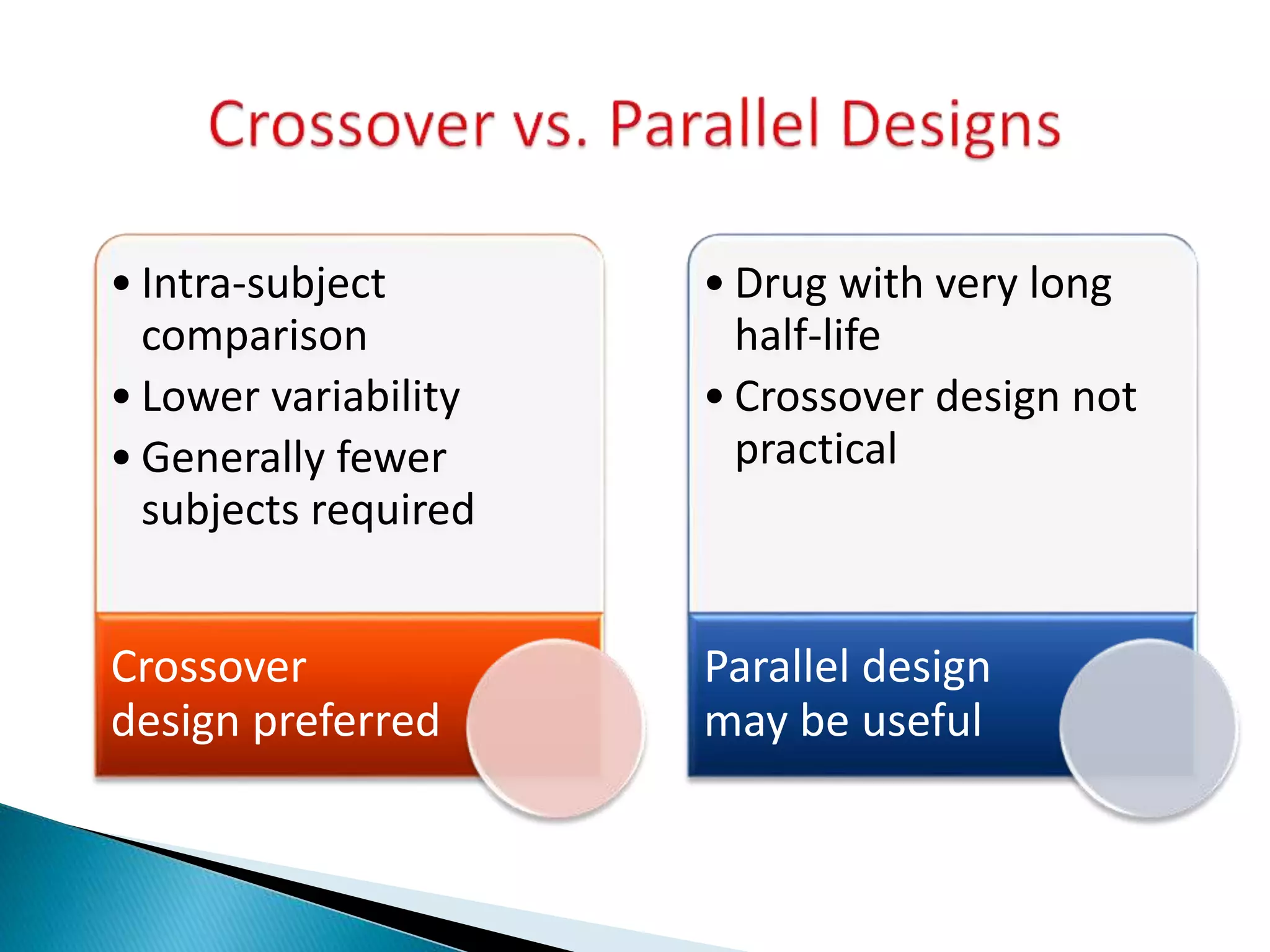








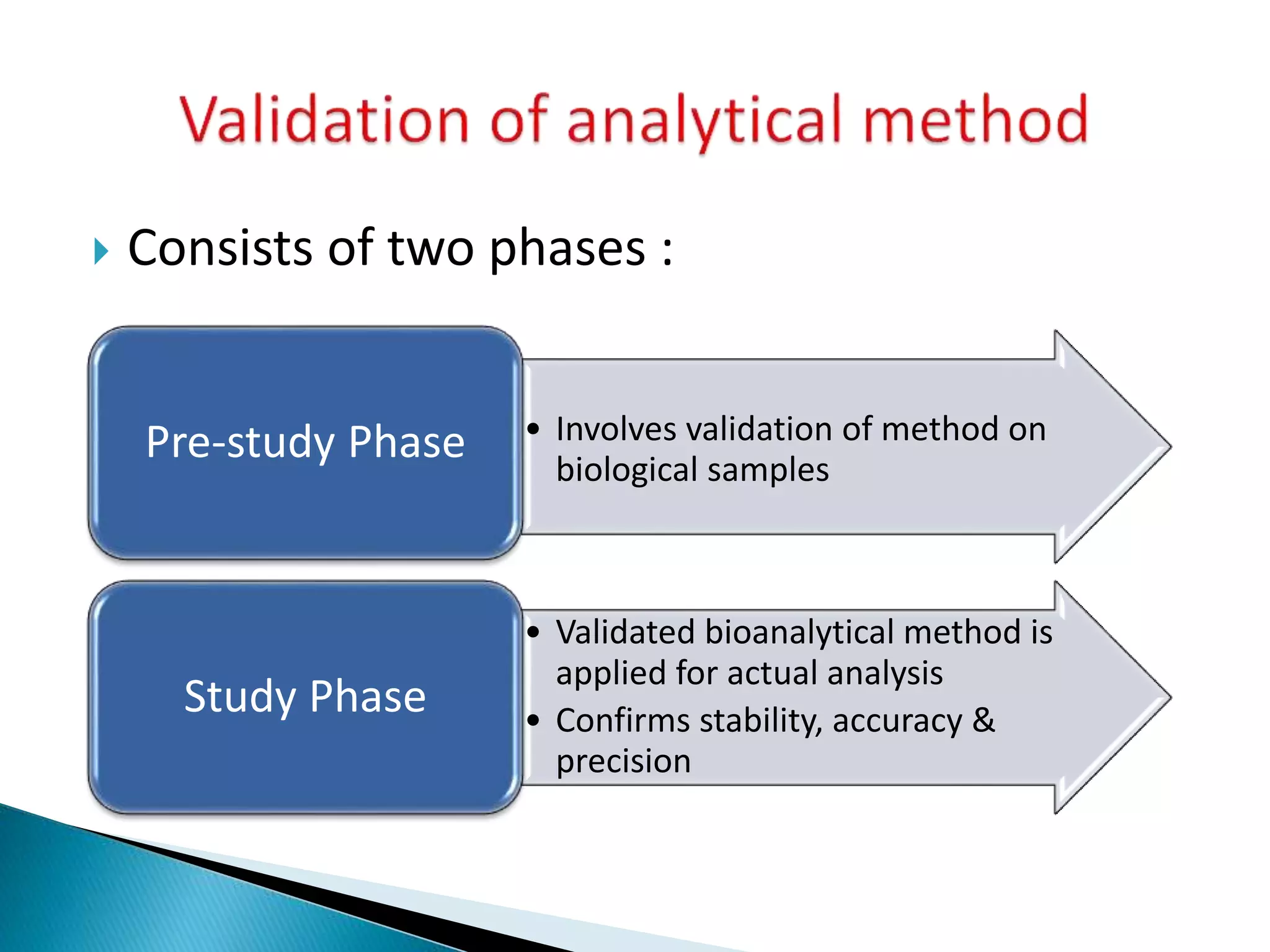










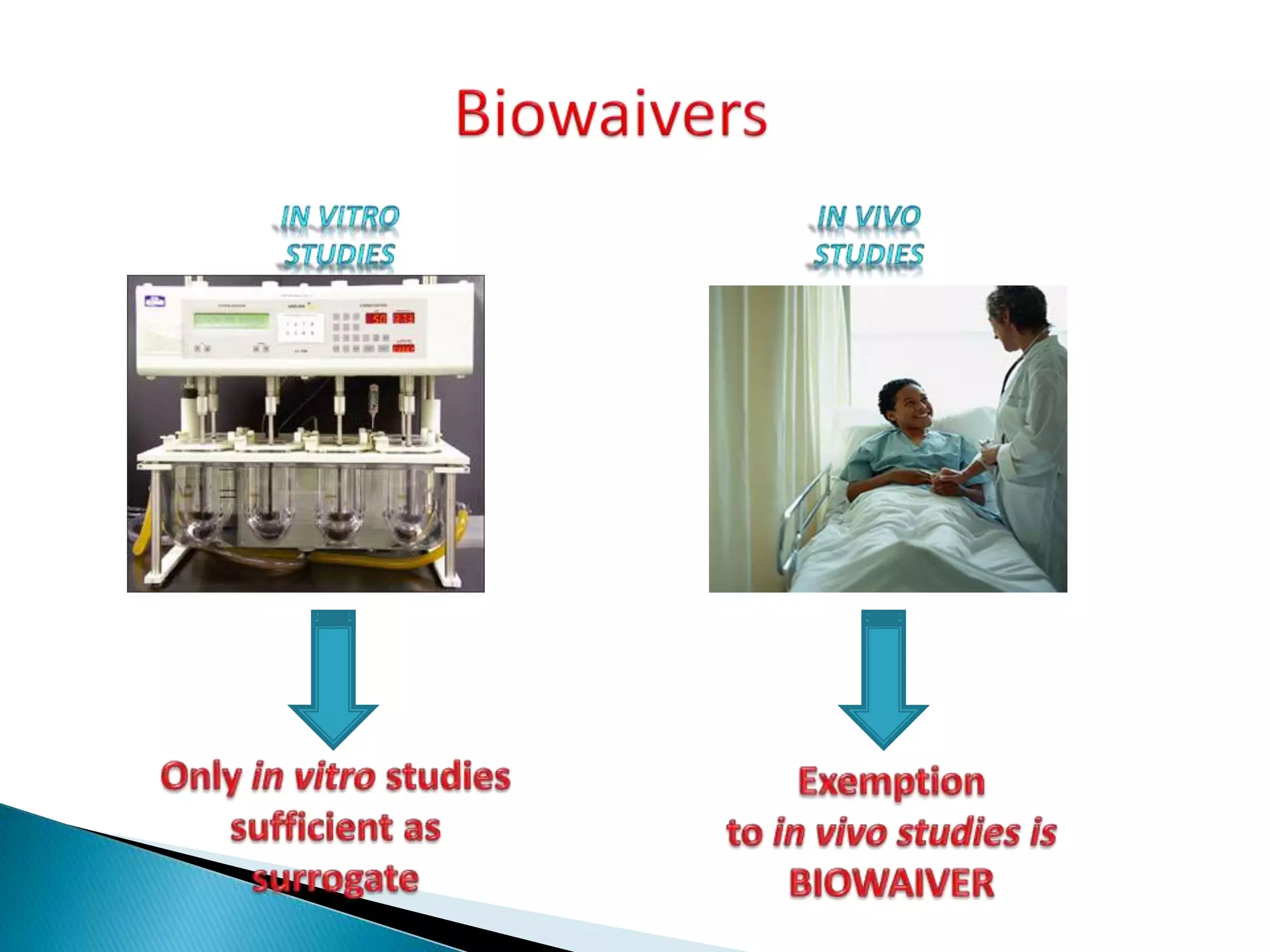



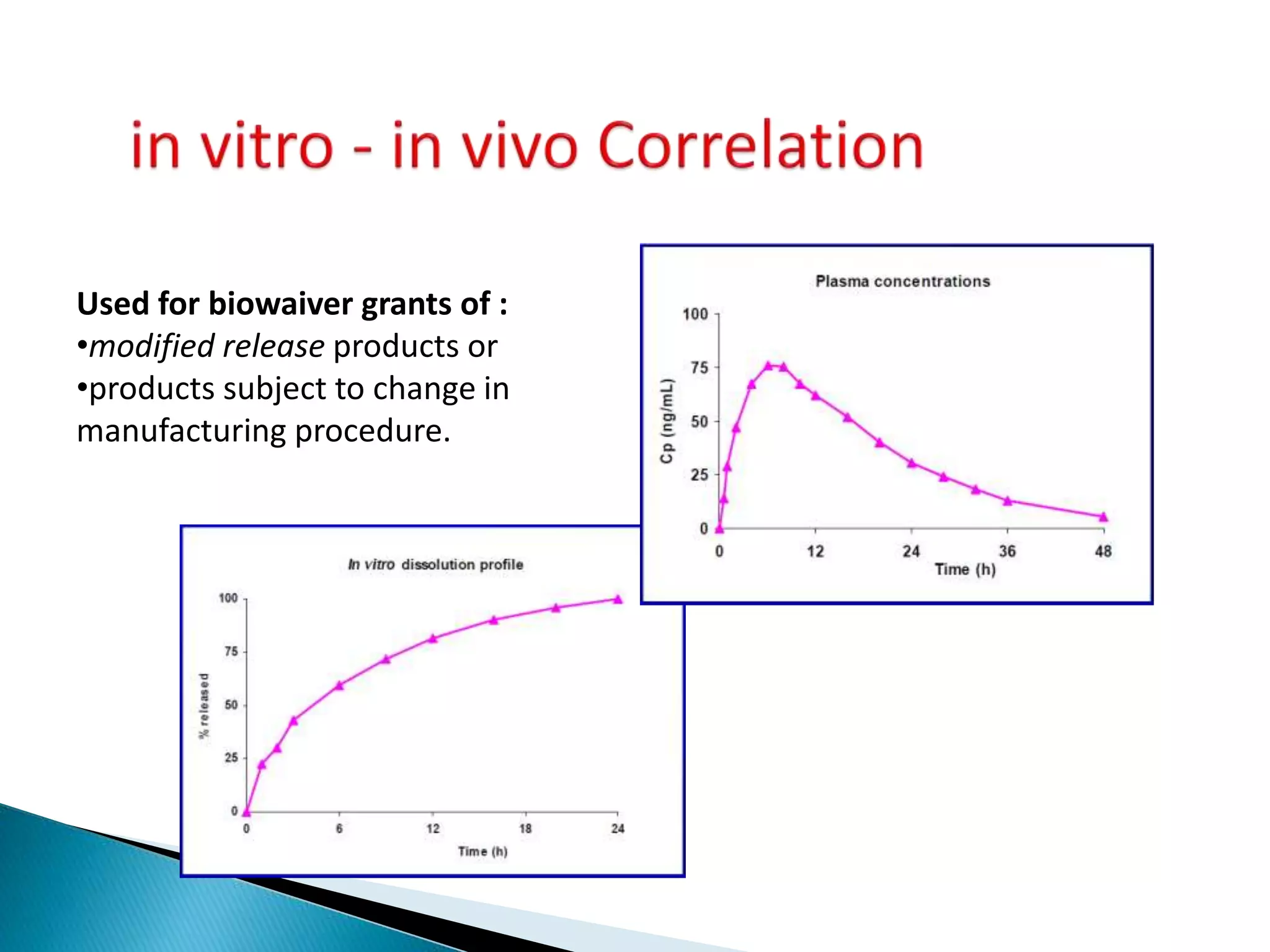



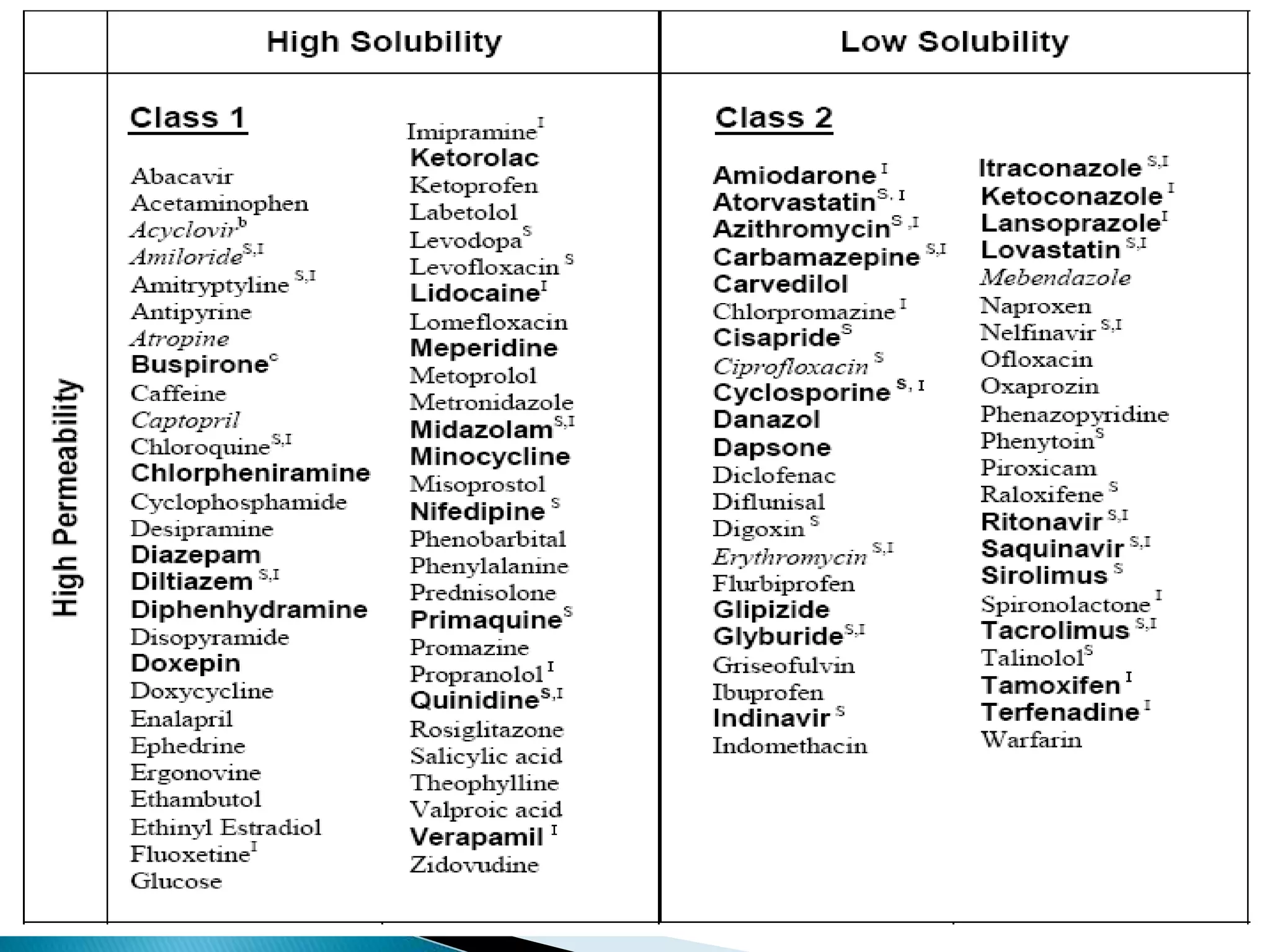
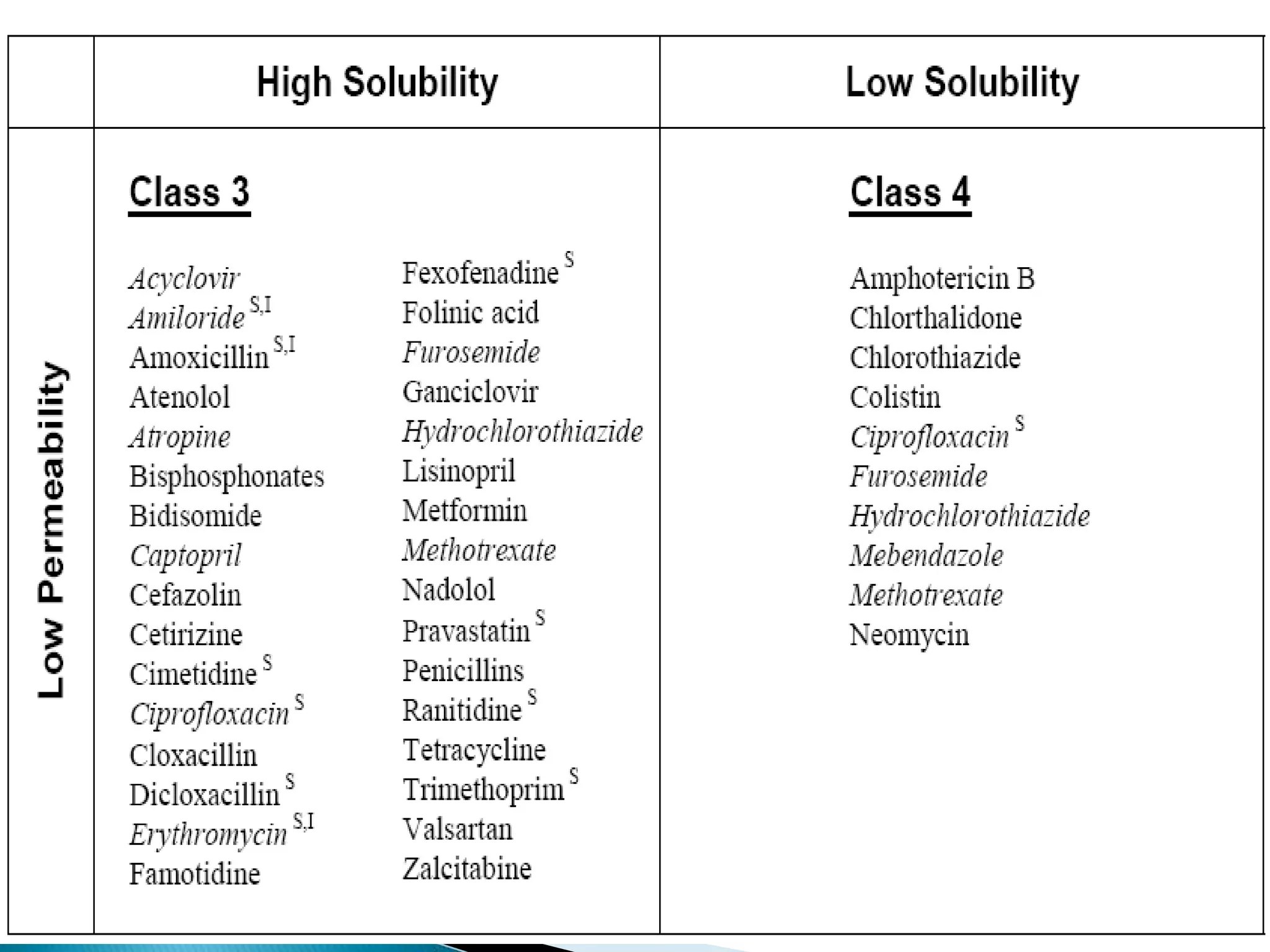






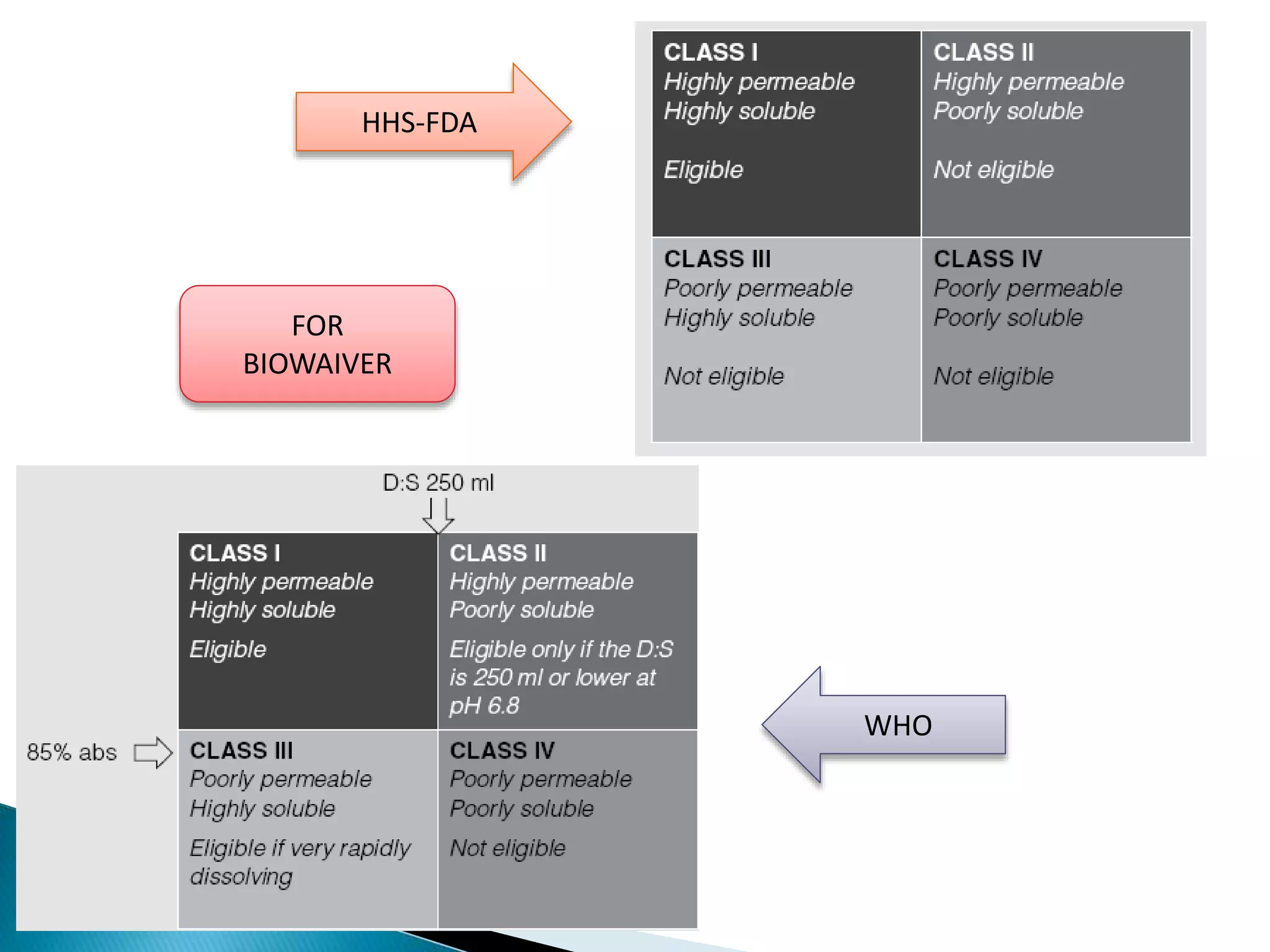












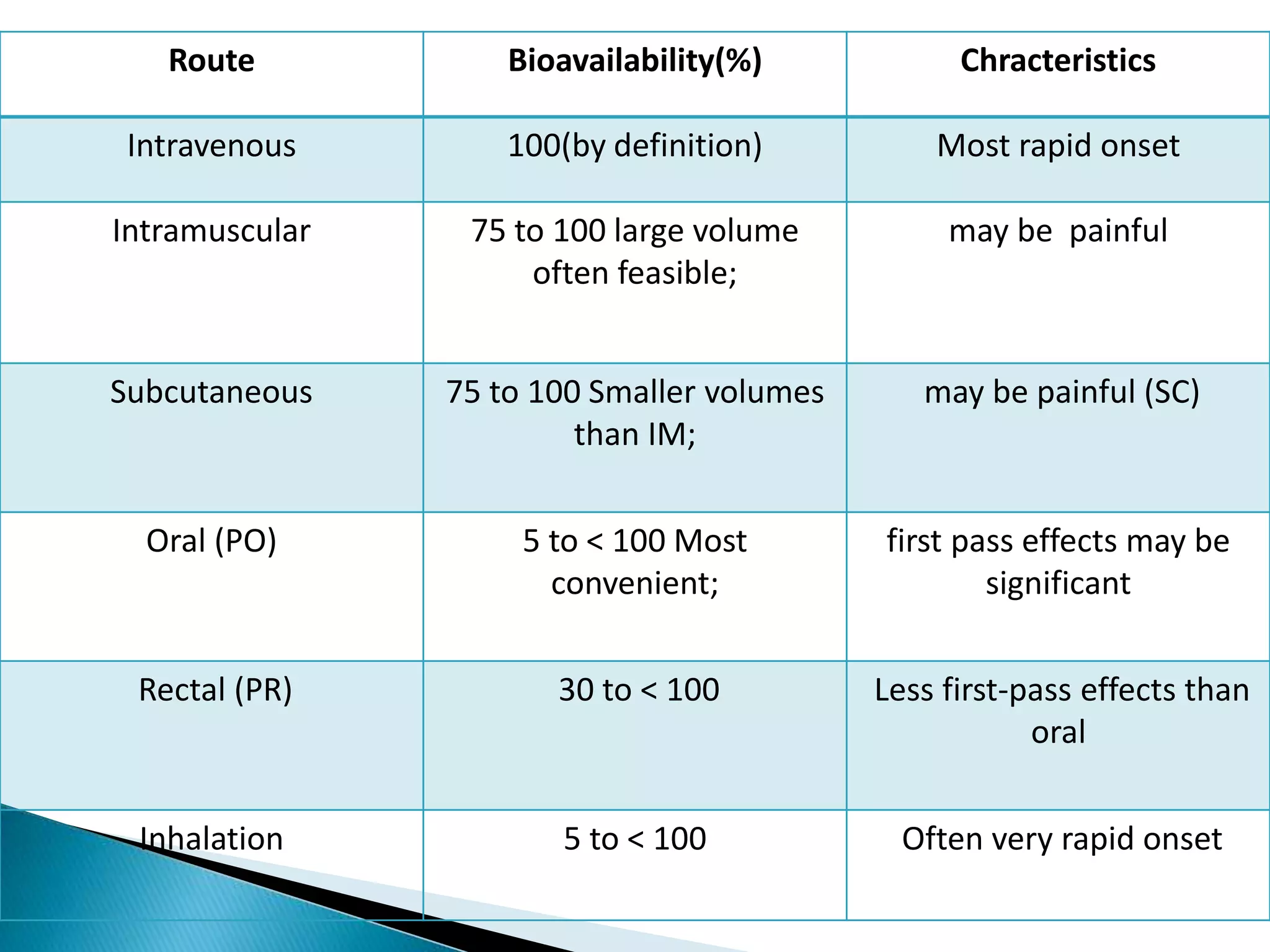
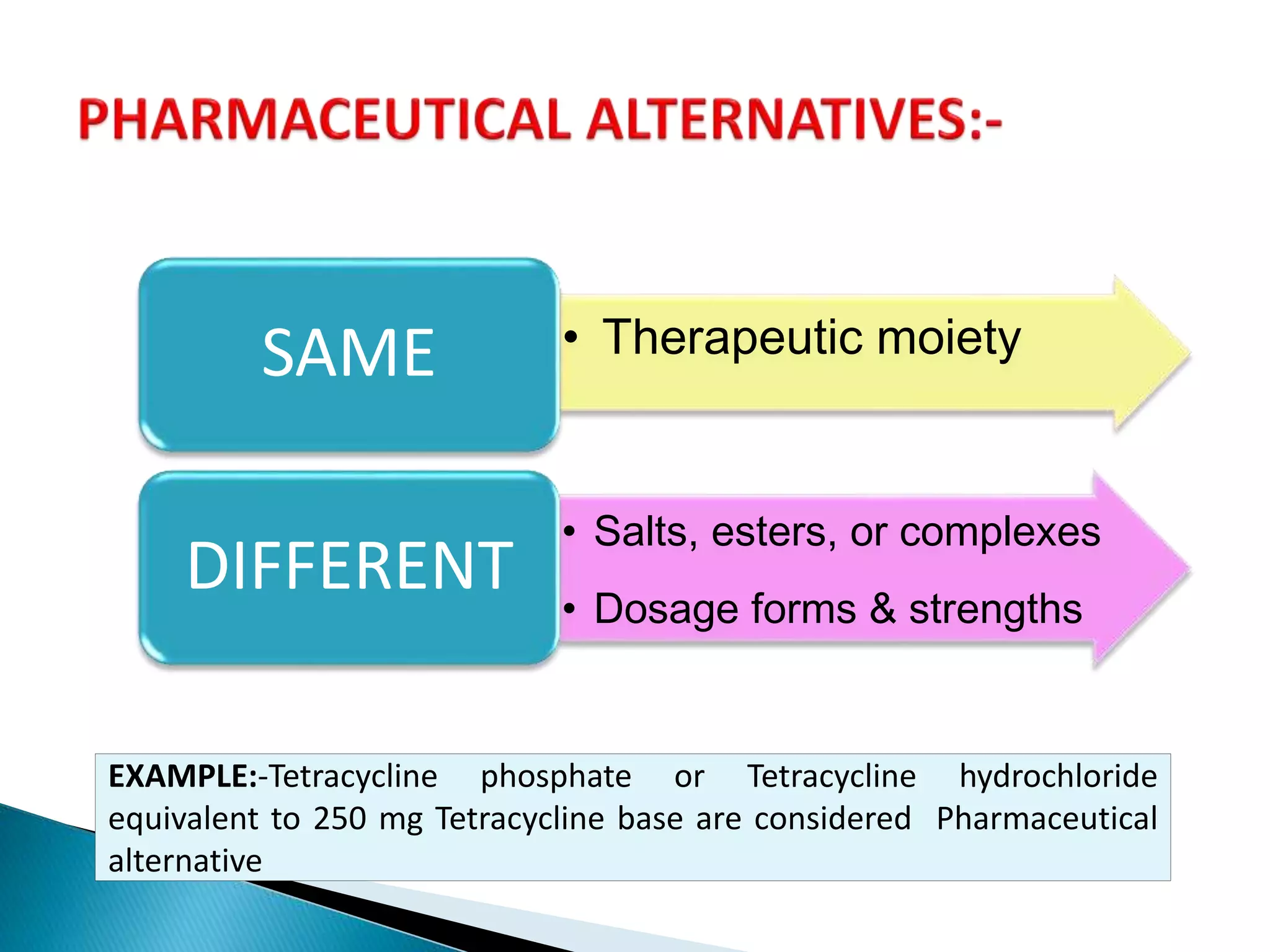
The document discusses the definitions and assessment methods for bioavailability and bioequivalence of drug formulations, emphasizing the importance of various drug administration routes, pharmacokinetics, and study design in evaluating drug effectiveness and safety. It outlines criteria for pharmaceutical and therapeutic equivalence and explores factors that influence study parameters, including subject population and sample size determination. The document also compares different study designs, such as cross-over and parallel designs, while emphasizing the control of variability and statistical considerations in bioequivalence studies.









![Abbreviated New Drug Application [ANDA]](https://cdn.slidesharecdn.com/ss_thumbnails/abbreviatednewdrugapplicationanda-160619062810-thumbnail.jpg?width=640&height=640&fit=bounds)











![Investigational New drug application [INDA]](https://cdn.slidesharecdn.com/ss_thumbnails/investigationalnewdrugapplicationinda-160619063044-thumbnail.jpg?width=640&height=640&fit=bounds)