Downloaded 645 times









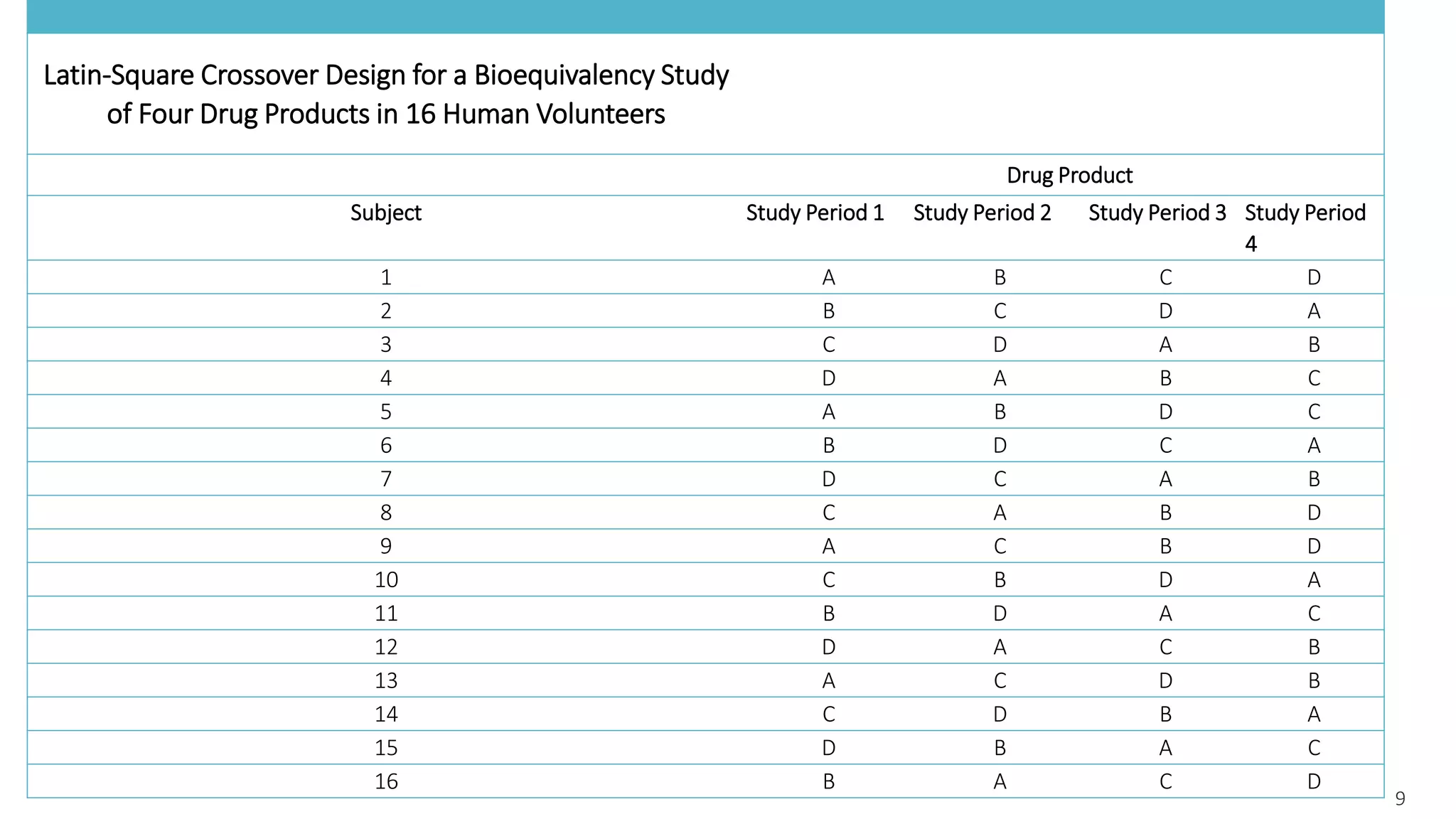


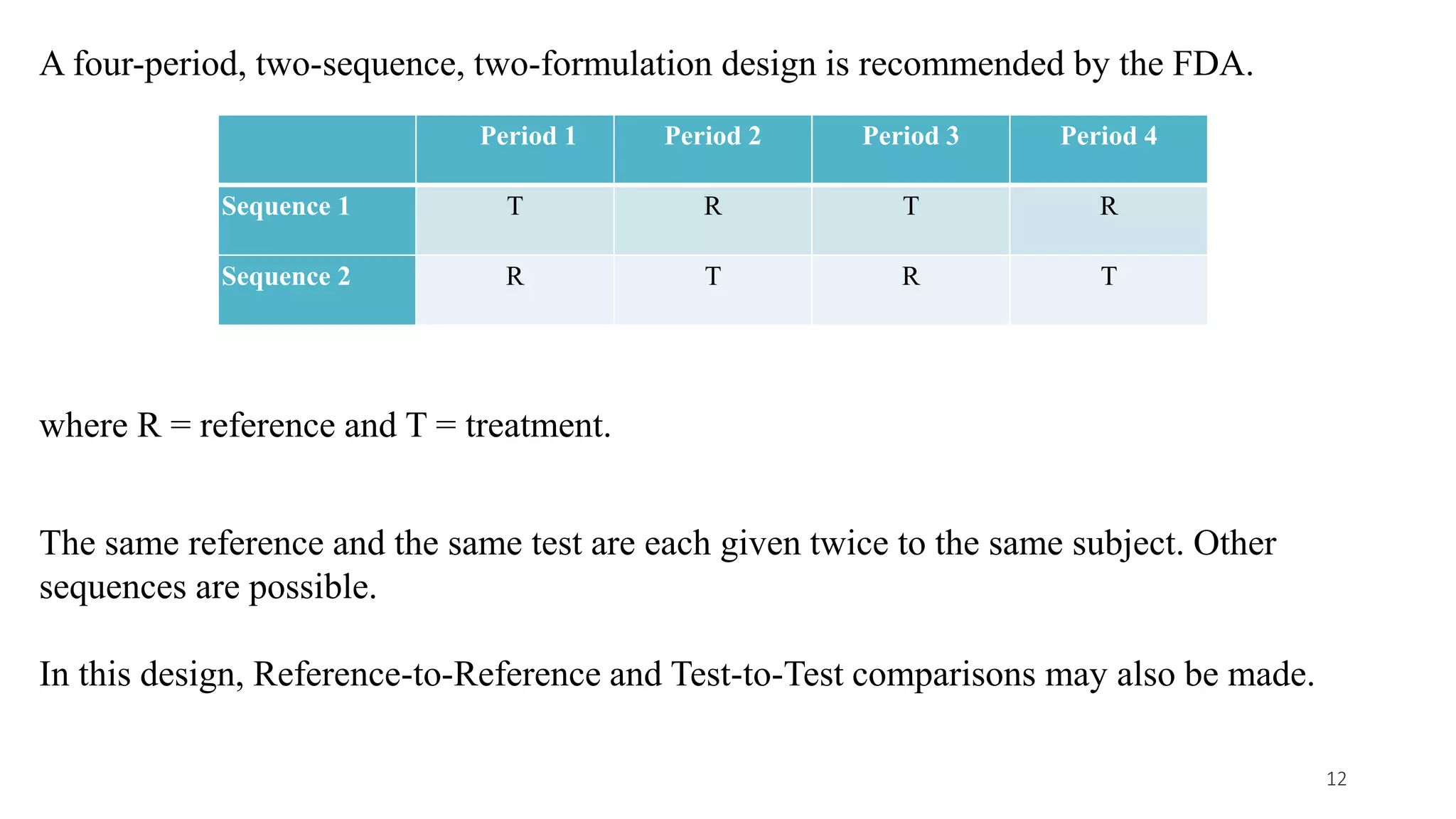
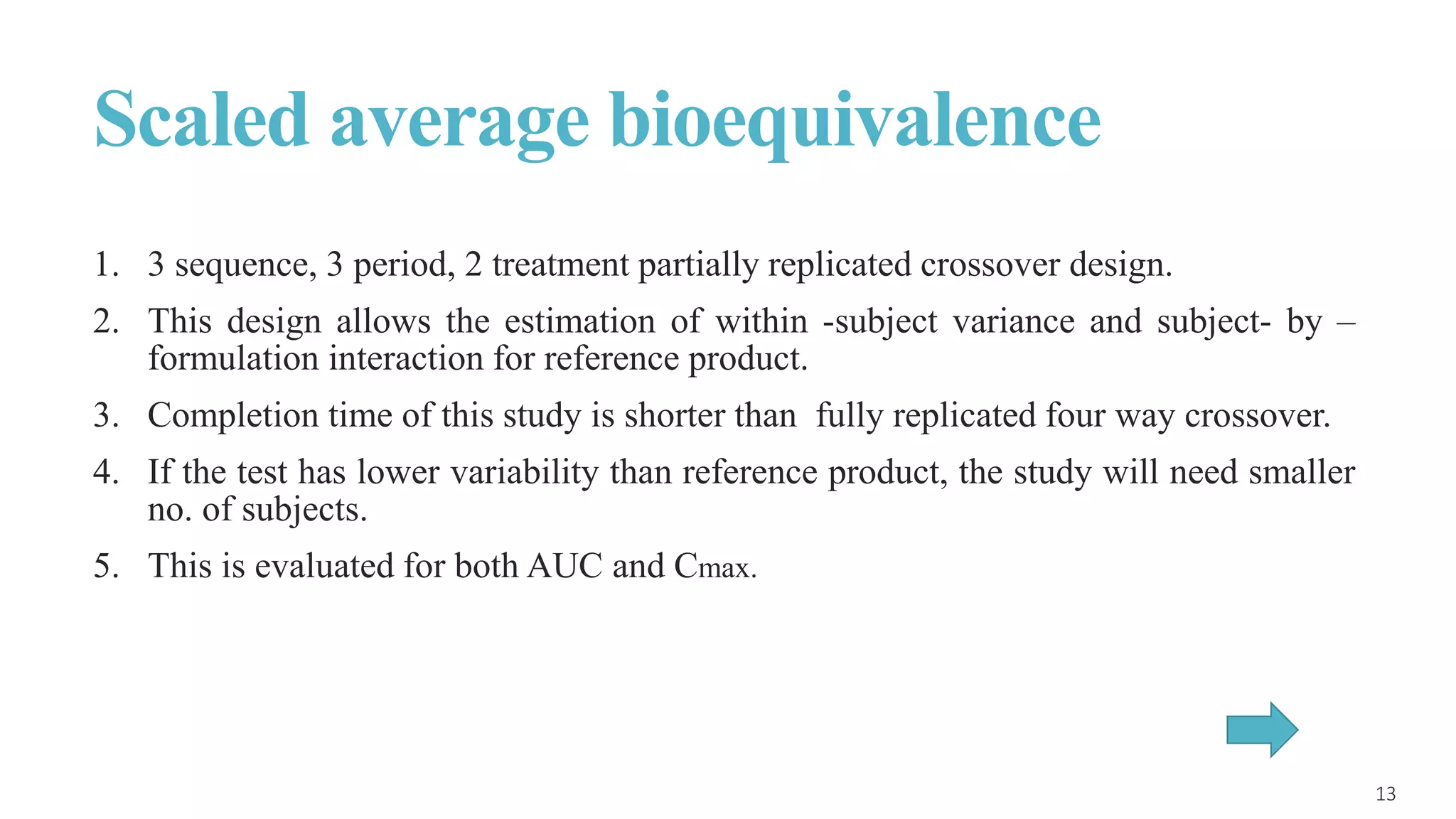










This document discusses various study designs used in bioequivalence studies. It describes the objectives of such studies as determining if a test product is bioequivalent to a reference product when administered at the same molar dose under similar conditions. Some key study designs discussed include non-replicated parallel studies for long-acting drugs, multiple dose steady state studies, and clinical endpoint studies comparing therapeutic effects. Special considerations are also noted for highly variable drugs.



































































