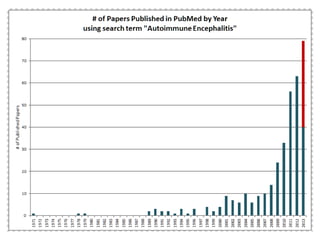
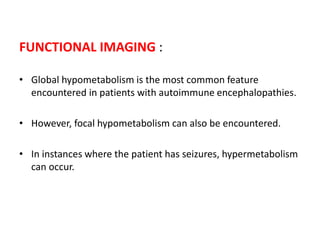
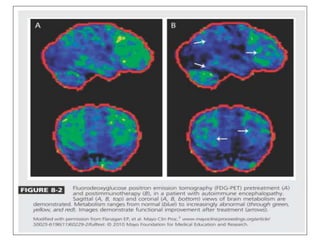
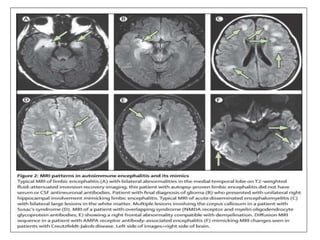
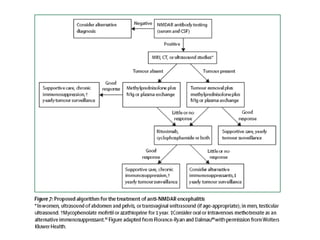
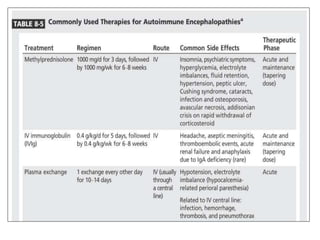
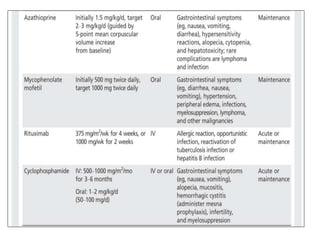
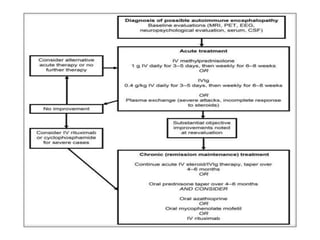
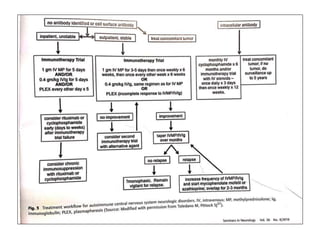
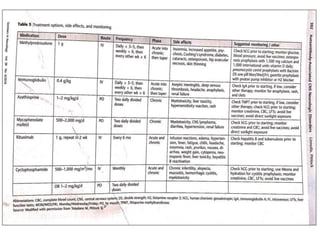
1) Autoimmune encephalitis is a debilitating neurological disorder caused by inflammation of the brain. It develops subacutely over weeks and can affect individuals of all ages.
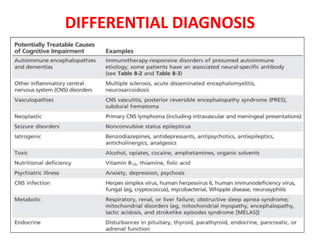
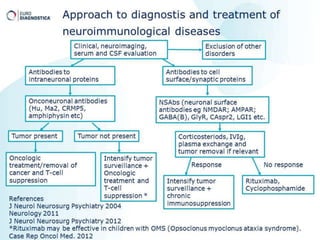
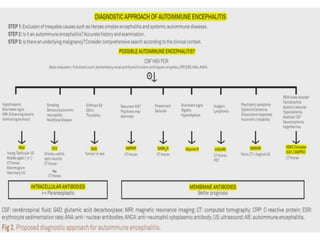
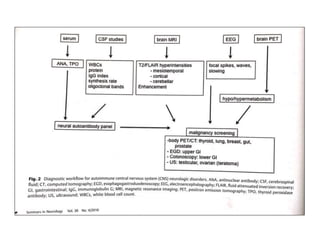
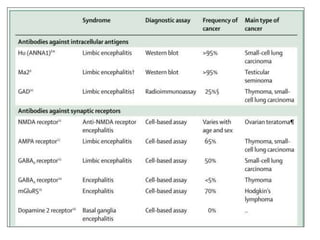
2) It has diverse clinical manifestations and immunological associations. Identification of neural autoantibodies has led to classification of different subtypes.
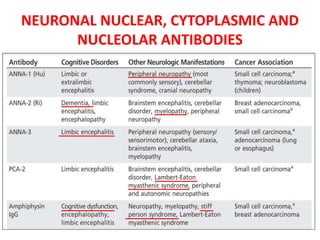
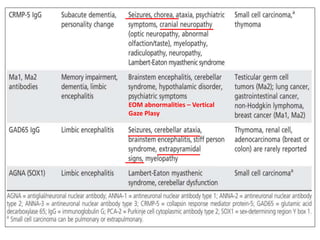
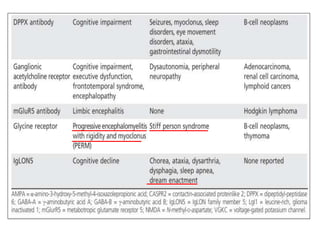
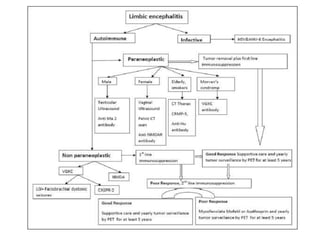
3) Prominent among these are anti-NMDAR encephalitis commonly seen in young women and children, autoimmune limbic encephalitis, and other syndromes associated with antibodies targeting neuronal cell-surface and intracellular antigens.
















































































![Myelin Oligodendrocyte Glycoprotein (MOG) Antibody Disease [MOG-AD]](https://cdn.slidesharecdn.com/ss_thumbnails/myelinoligodendrocyteglycoproteinmogantibodydisease0920-200920055159-thumbnail.jpg?width=640&height=640&fit=bounds)



































![DUAL AND TRIPLE ANTITHROMBOTIC THERAPY FOR SECONDARY STROKE [Autosaved].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/dualandtripleantithrombotictherapyforsecondarystrokeautosaved-230904113552-c3502b37-thumbnail.jpg?width=640&height=640&fit=bounds)

































