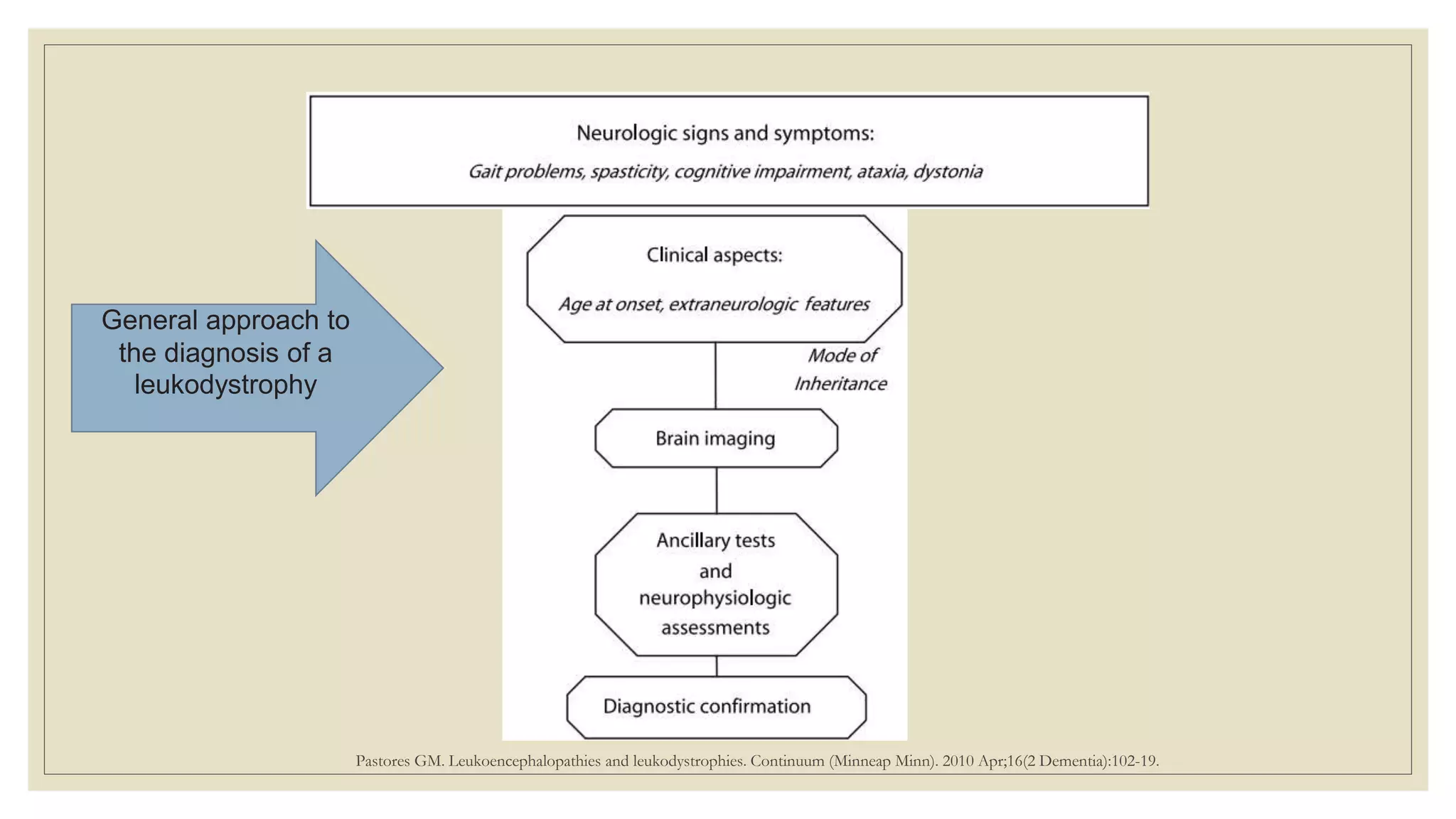
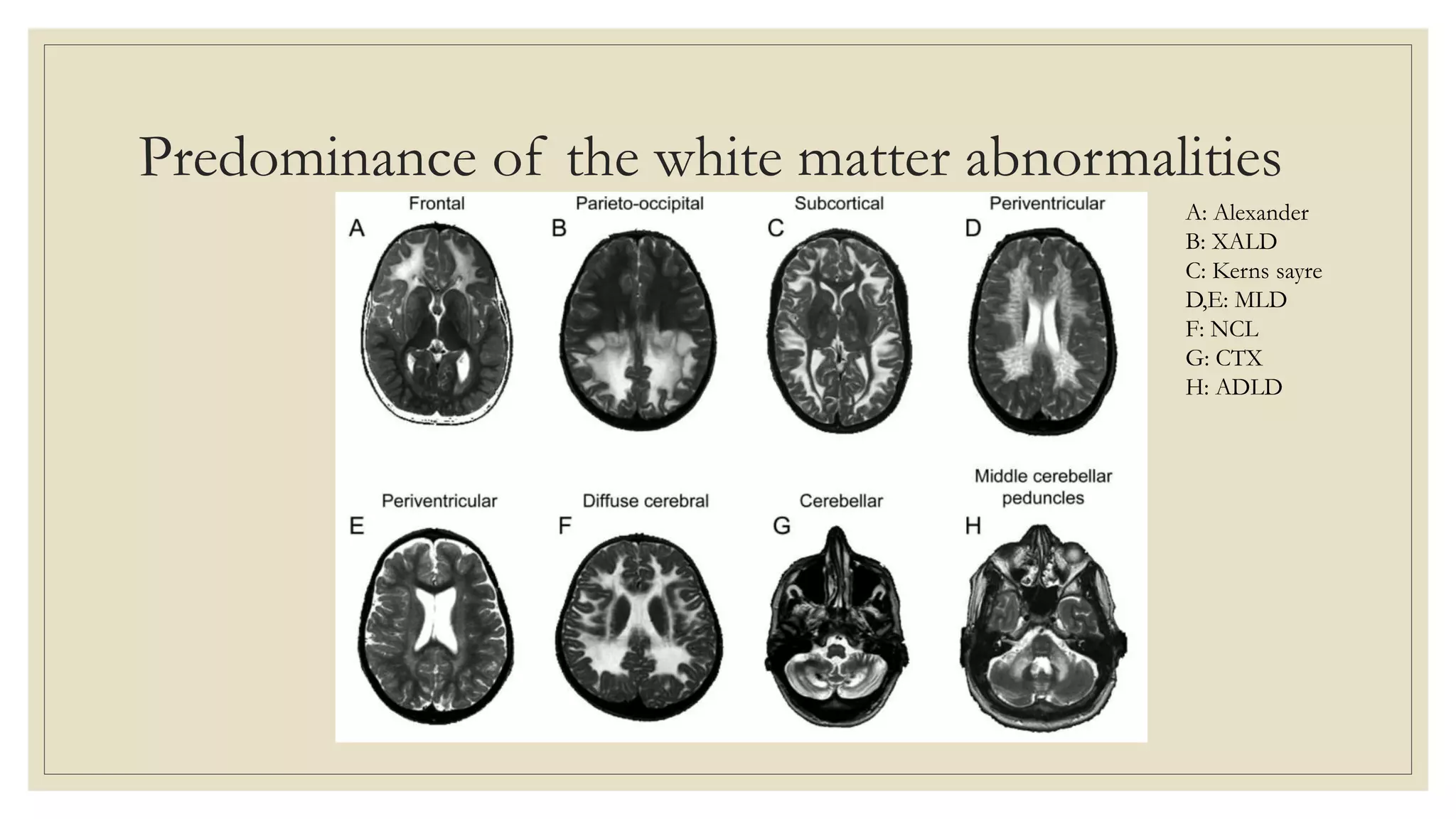
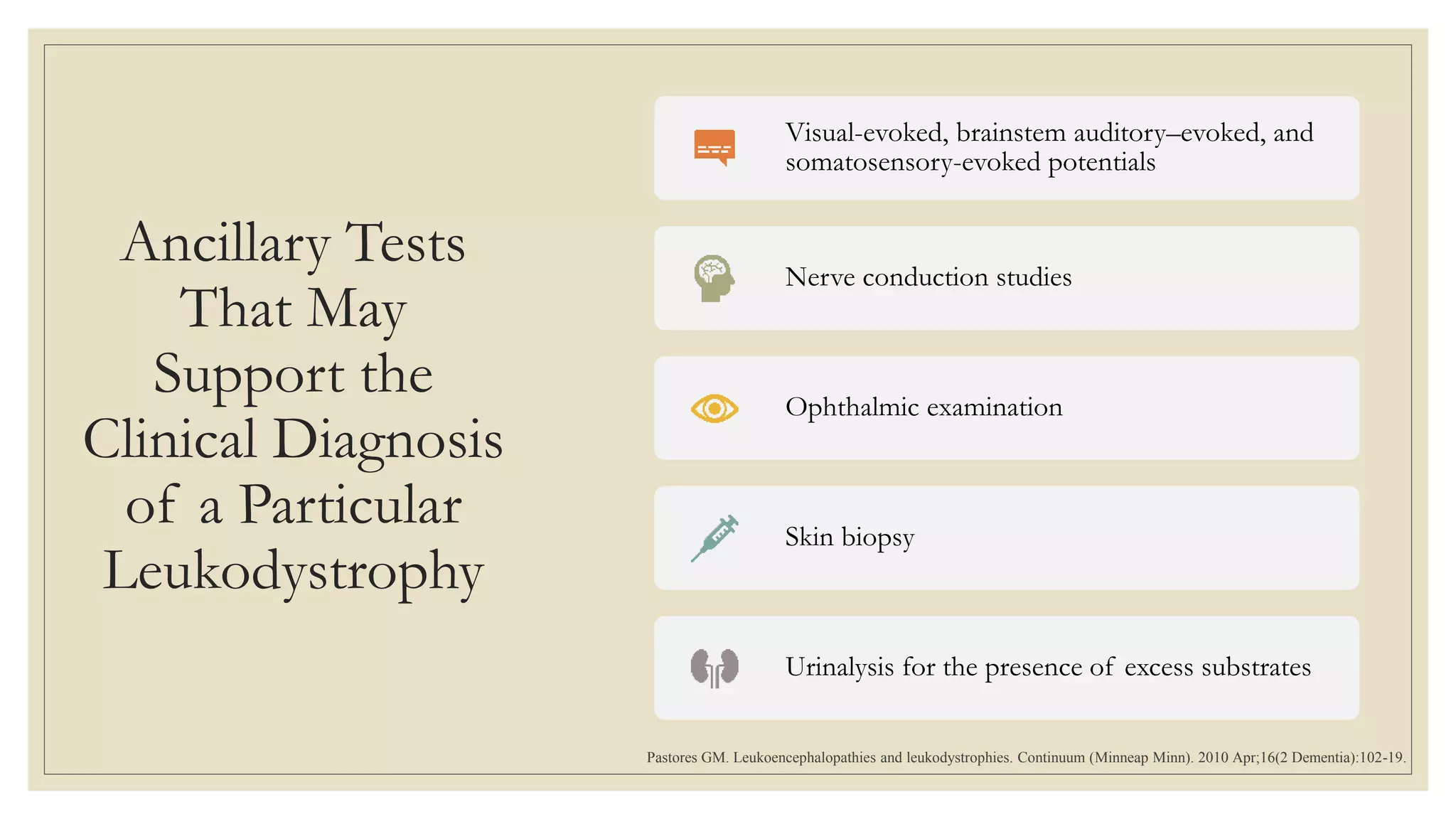
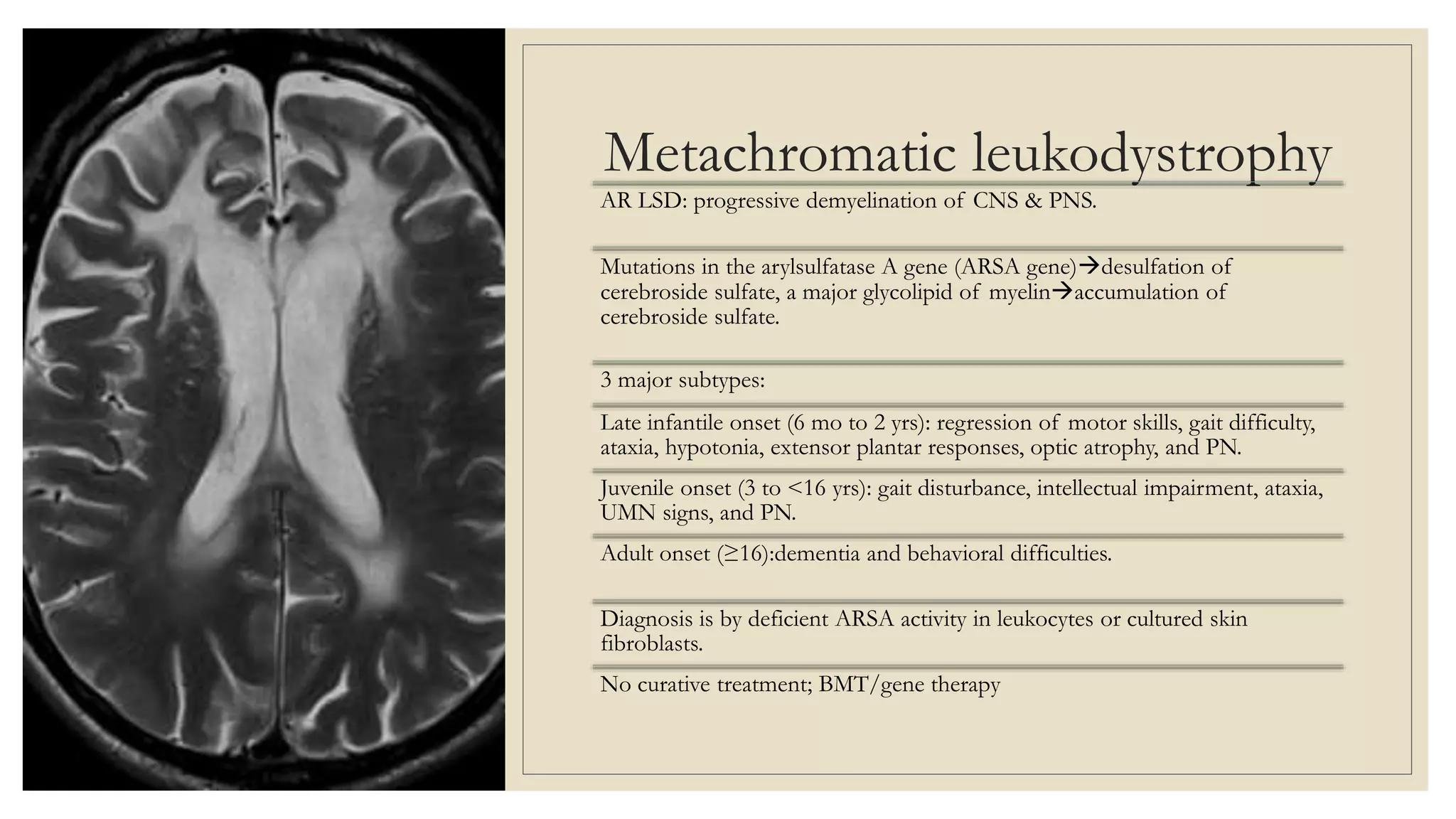
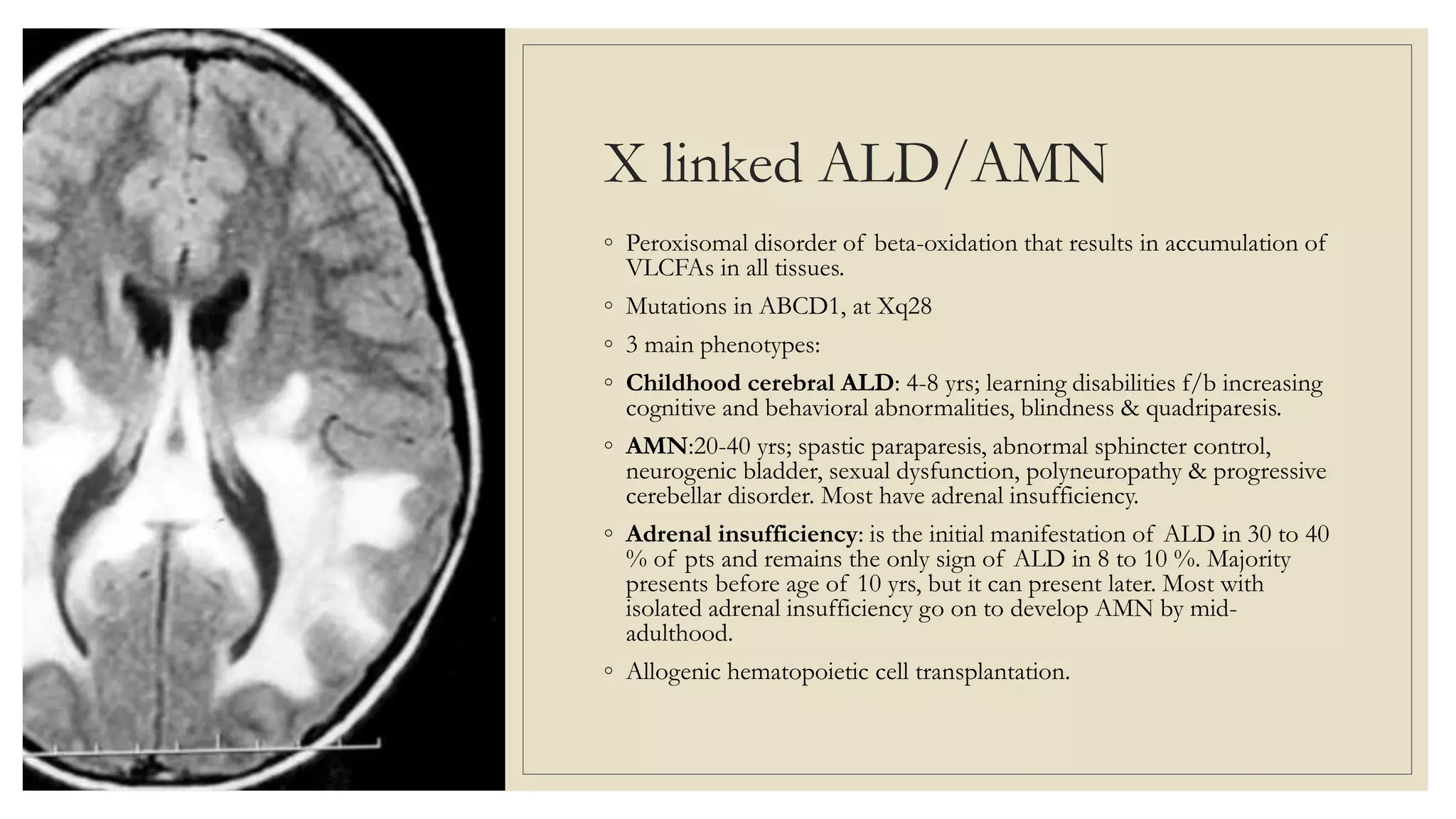
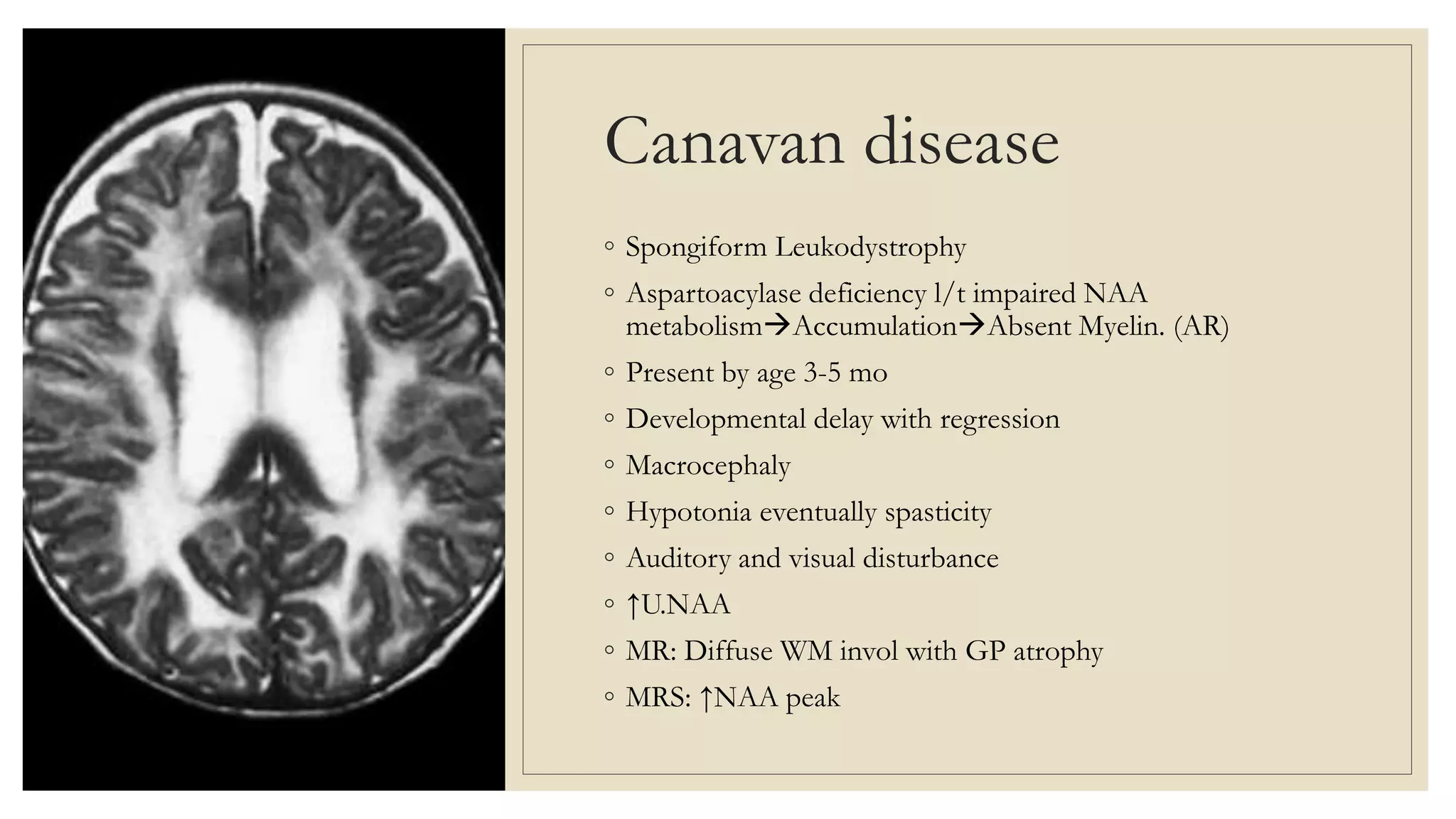
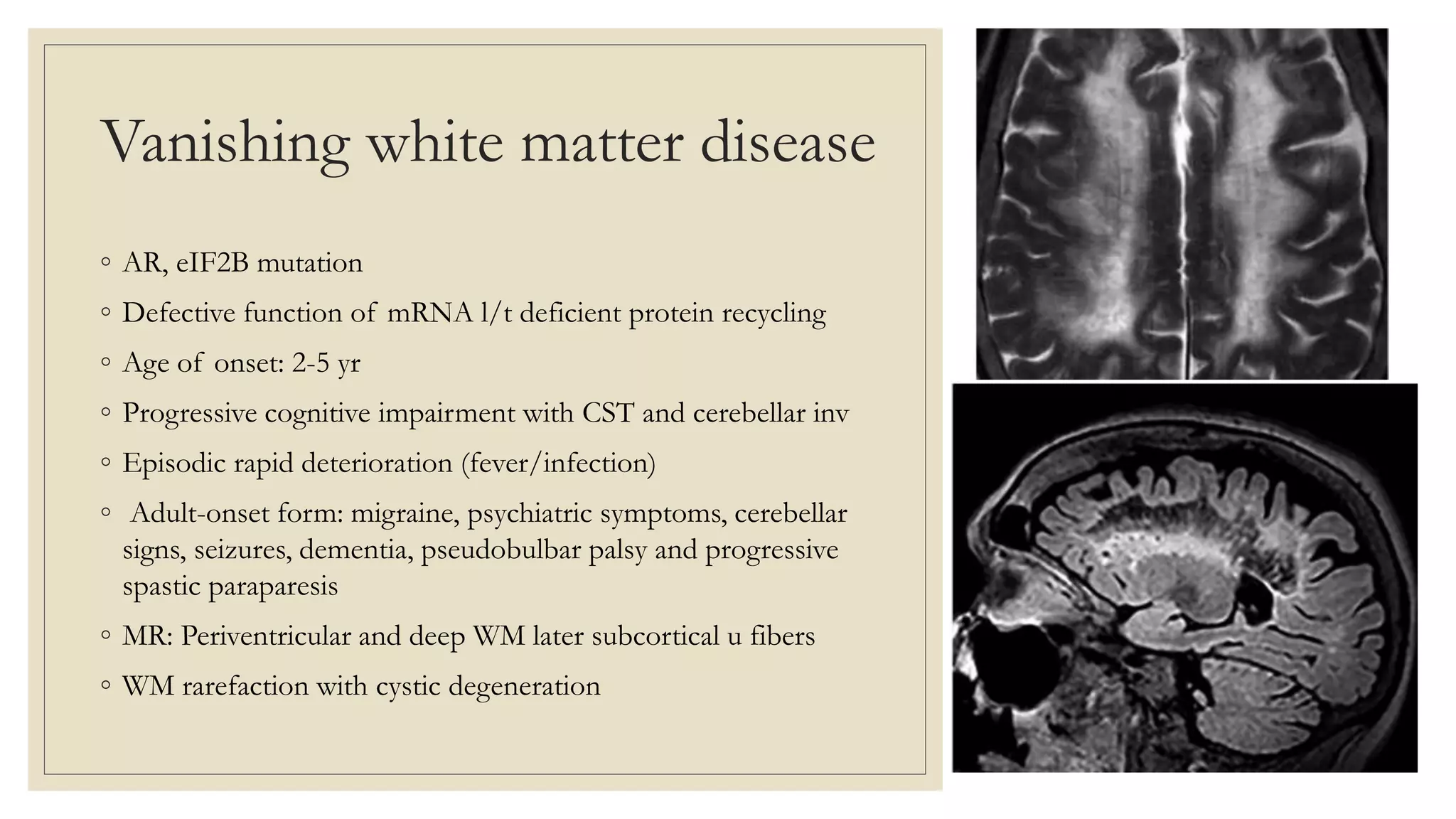
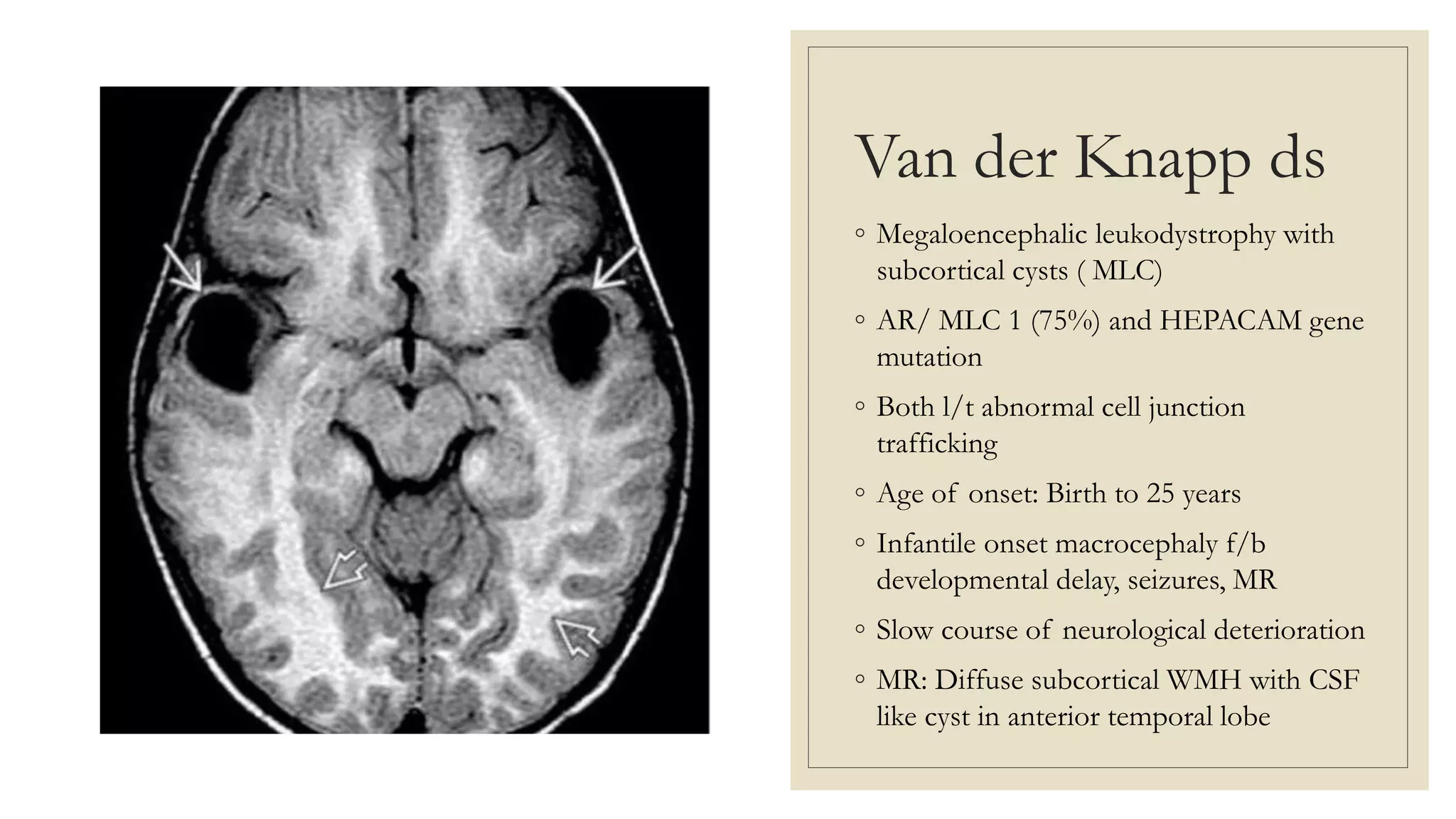
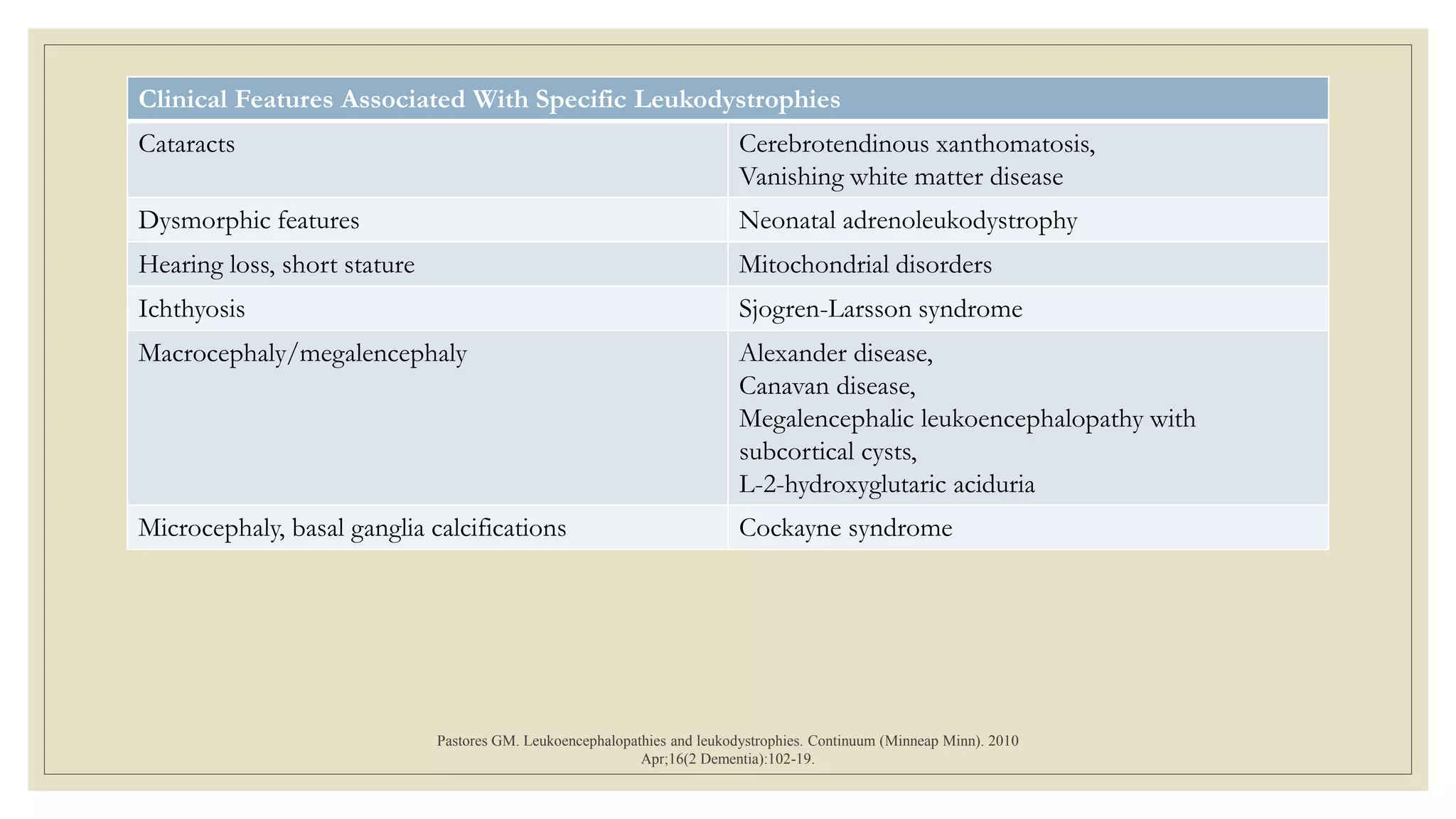
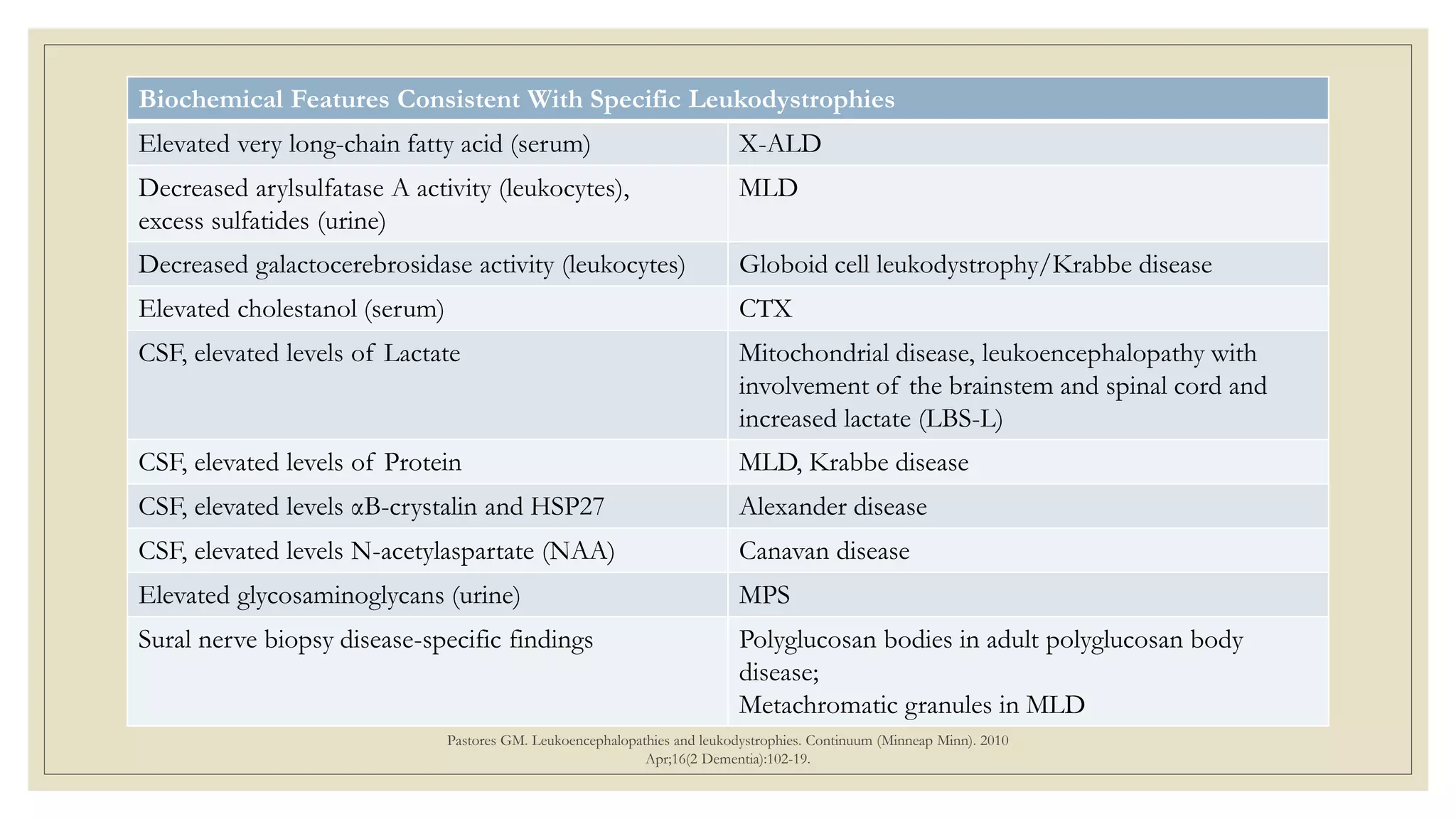
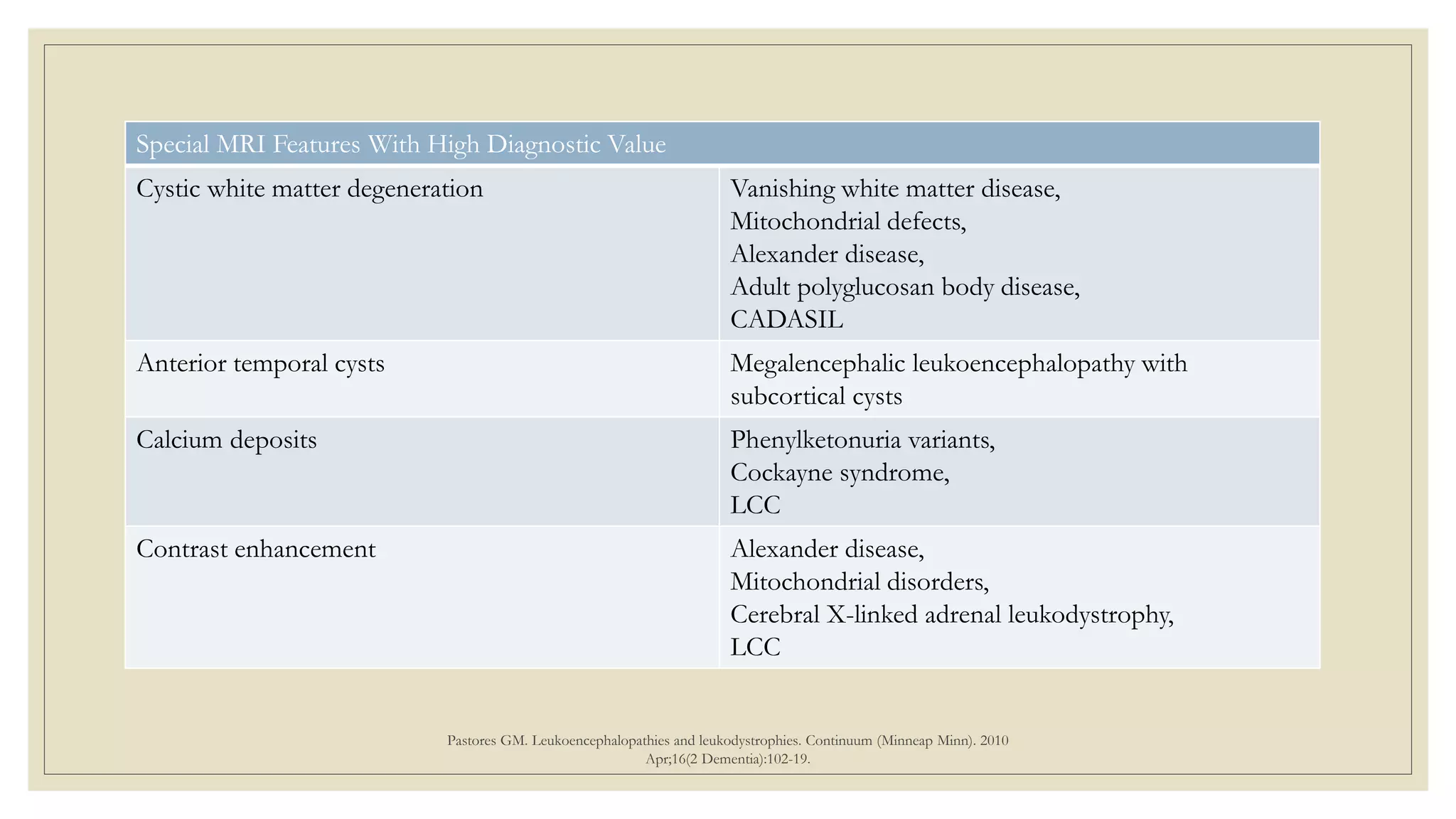
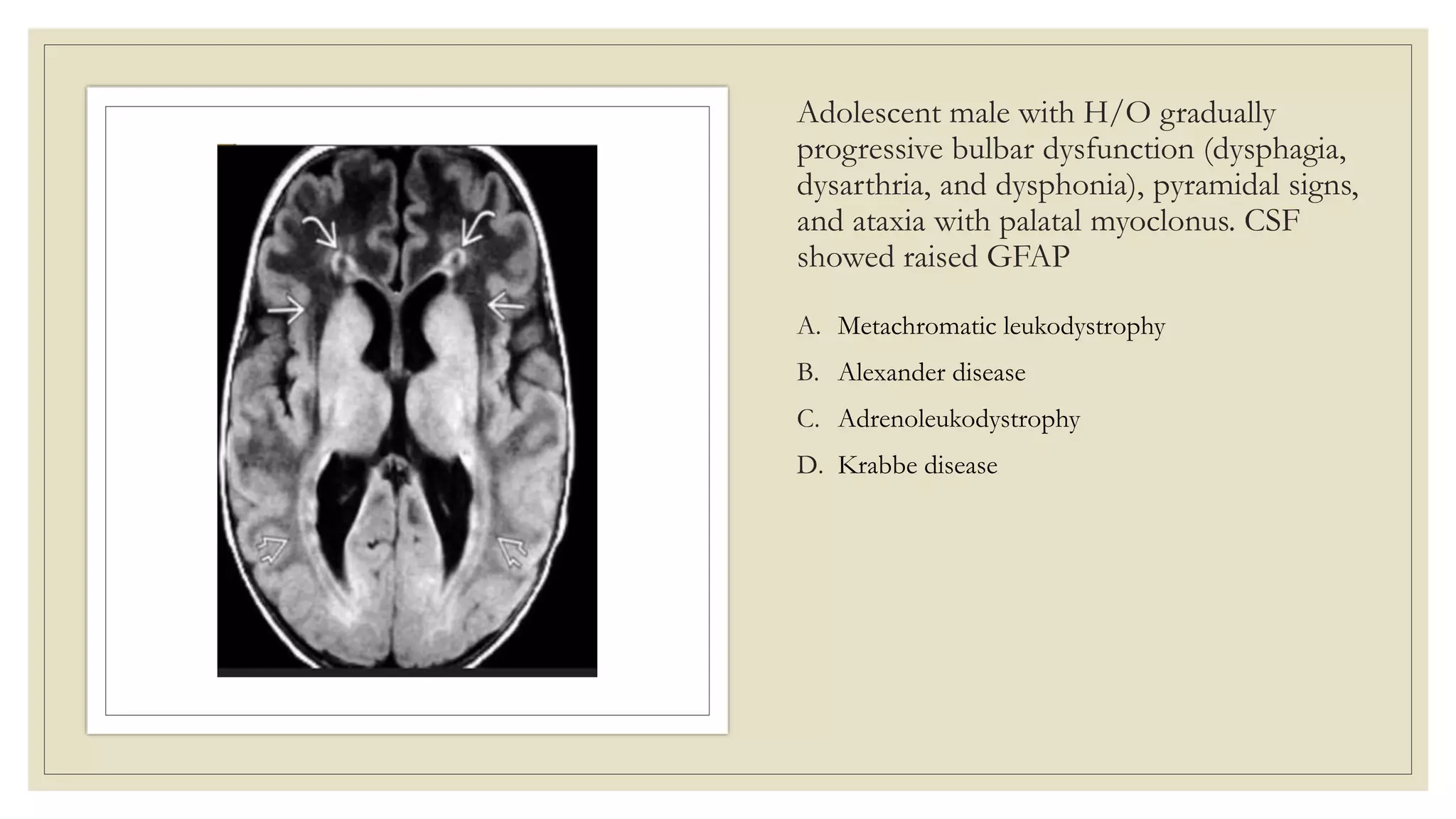
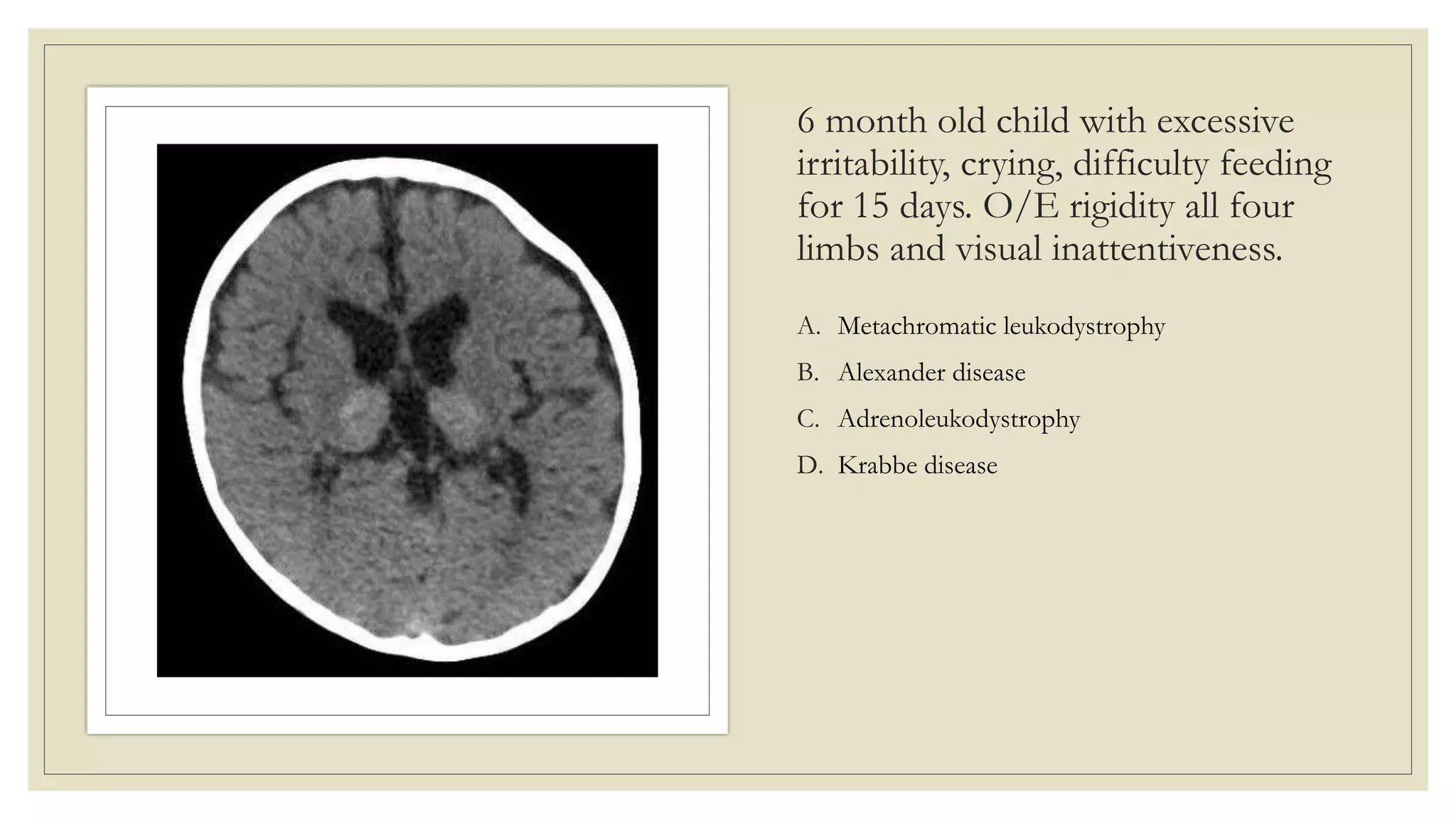
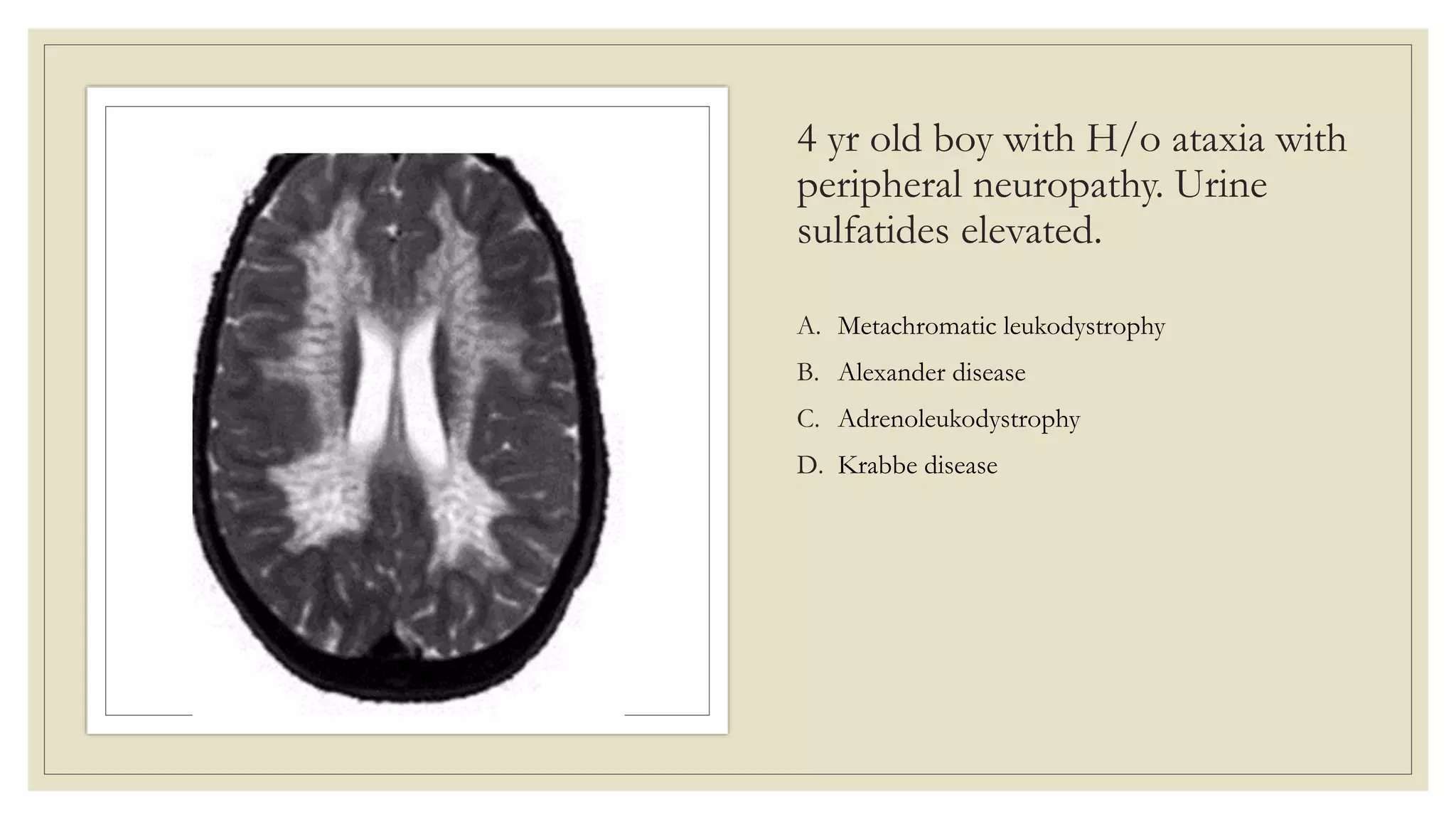
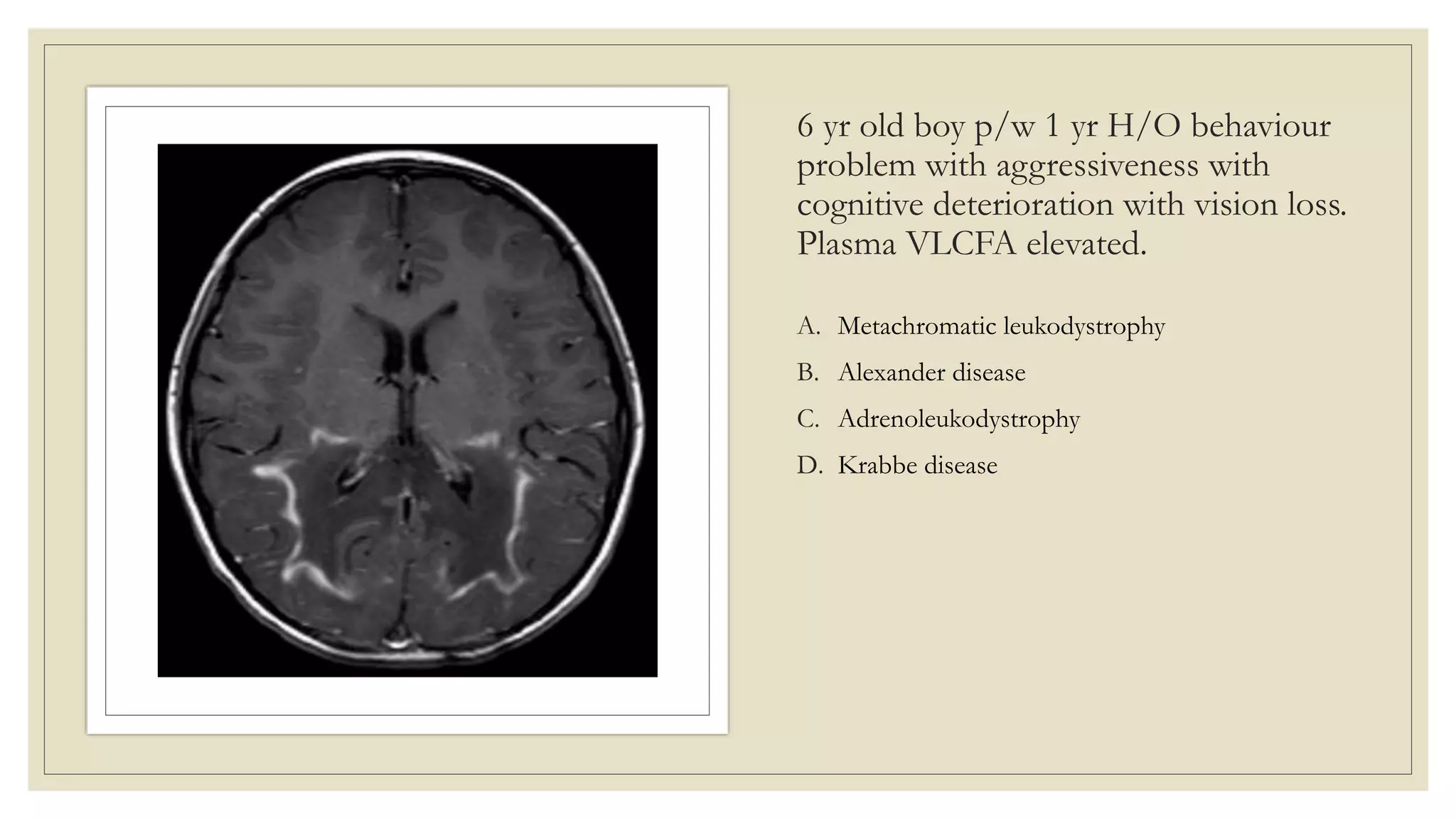
This document provides an overview of approaches to diagnosing leukodystrophies. It begins by defining leukodystrophies and differentiating them from other white matter disorders. Clinical features that suggest a leukodystrophy are described. A 3-step MRI approach is outlined involving identifying symmetric white matter involvement, patterns of involvement, and distinctive features. Common leukodystrophies in adults are discussed in detail including clinical presentation, genetics, imaging findings, and diagnostic testing. The document emphasizes a systematic approach to diagnosis utilizing clinical features, imaging, and ancillary tests.






















































































![DUAL AND TRIPLE ANTITHROMBOTIC THERAPY FOR SECONDARY STROKE [Autosaved].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/dualandtripleantithrombotictherapyforsecondarystrokeautosaved-230904113552-c3502b37-thumbnail.jpg?width=640&height=640&fit=bounds)

































