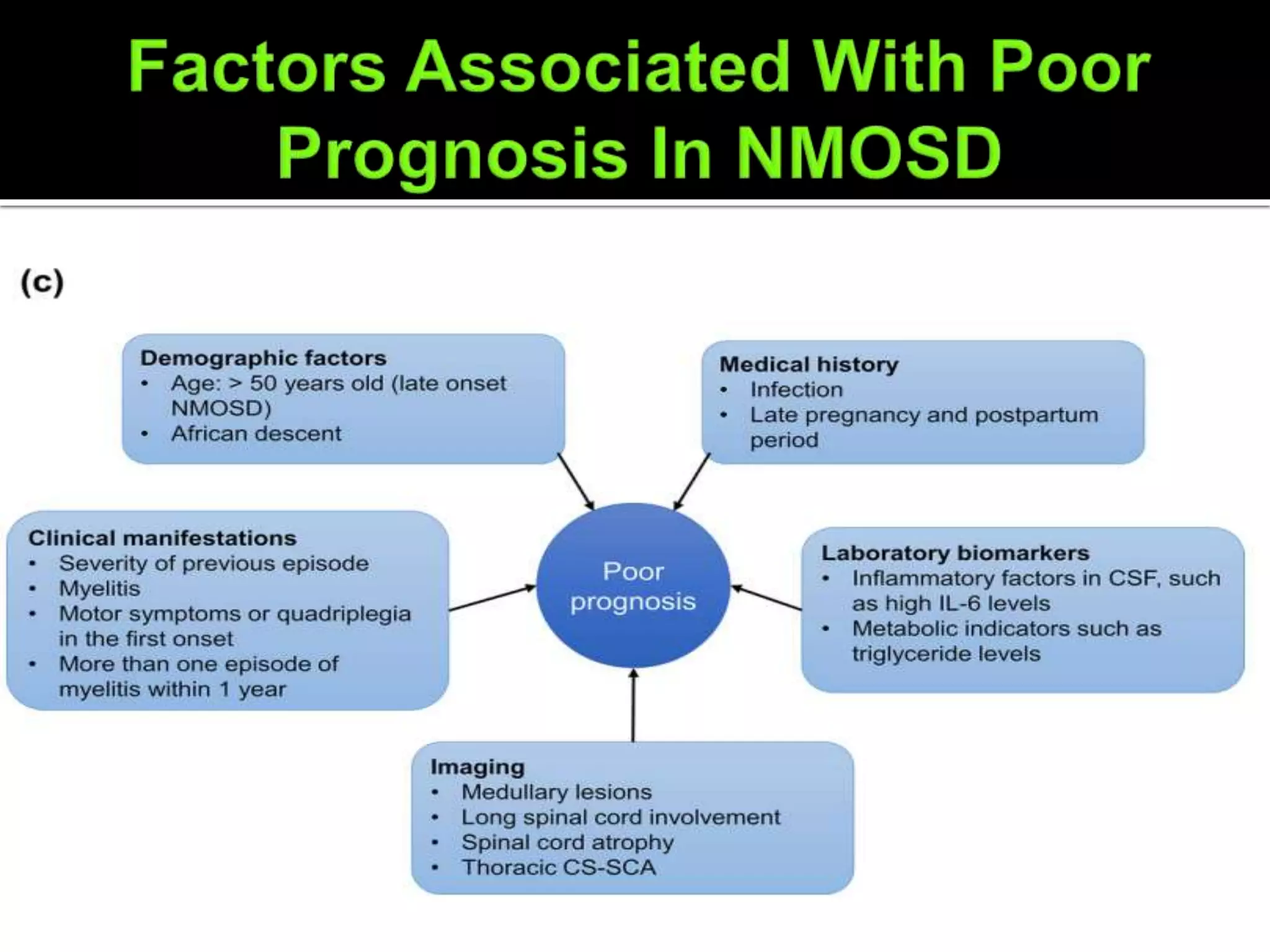
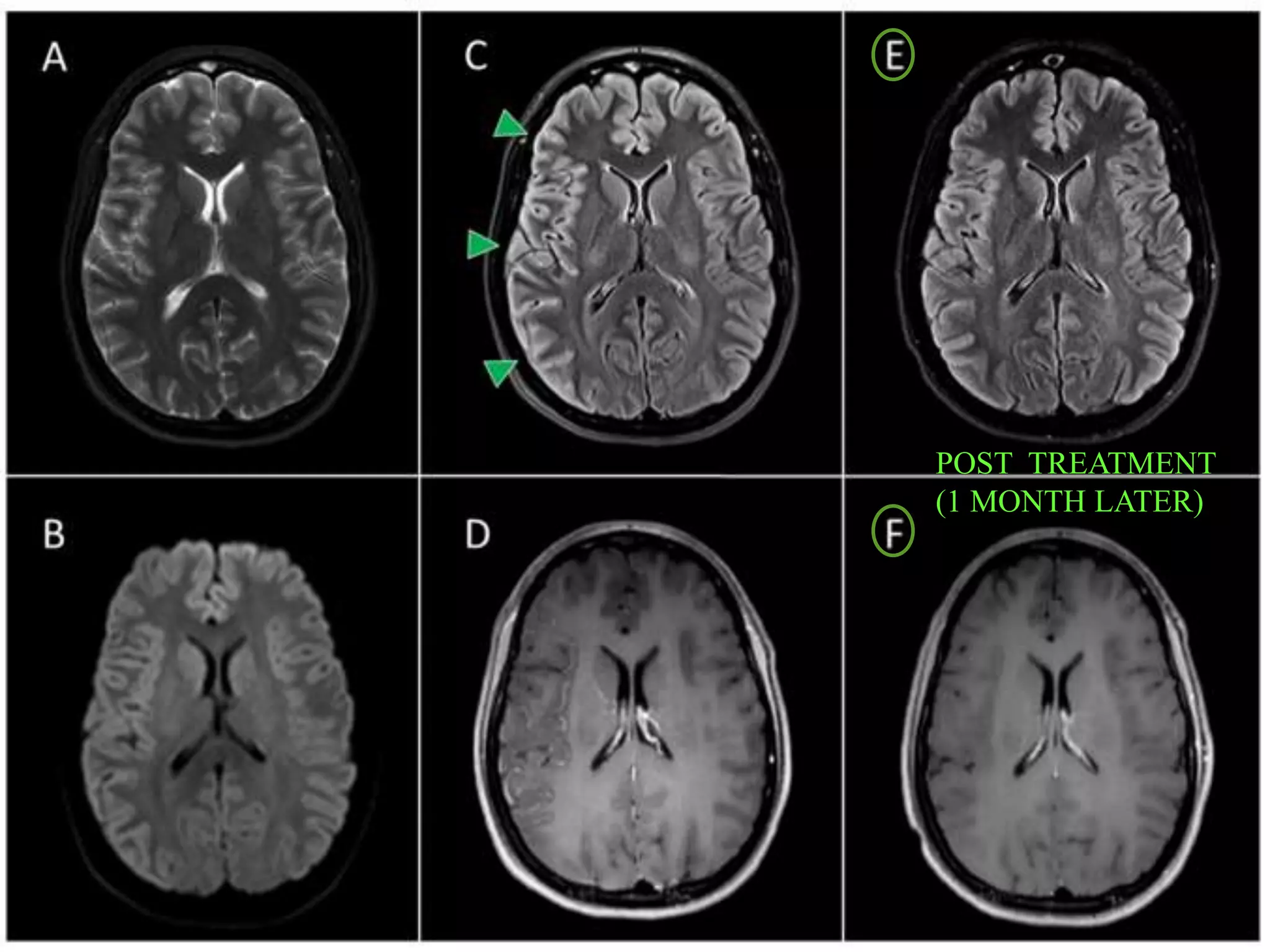
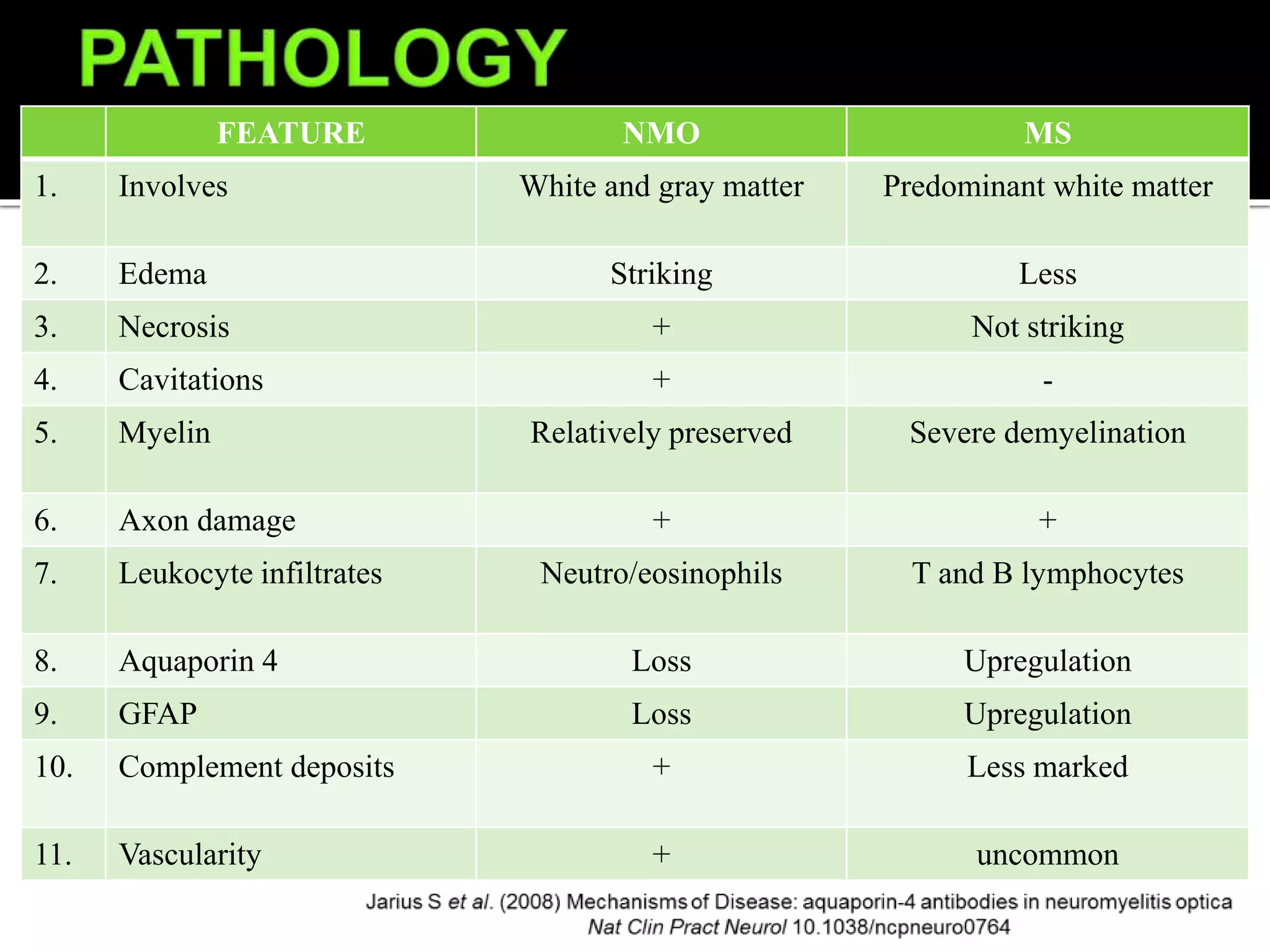
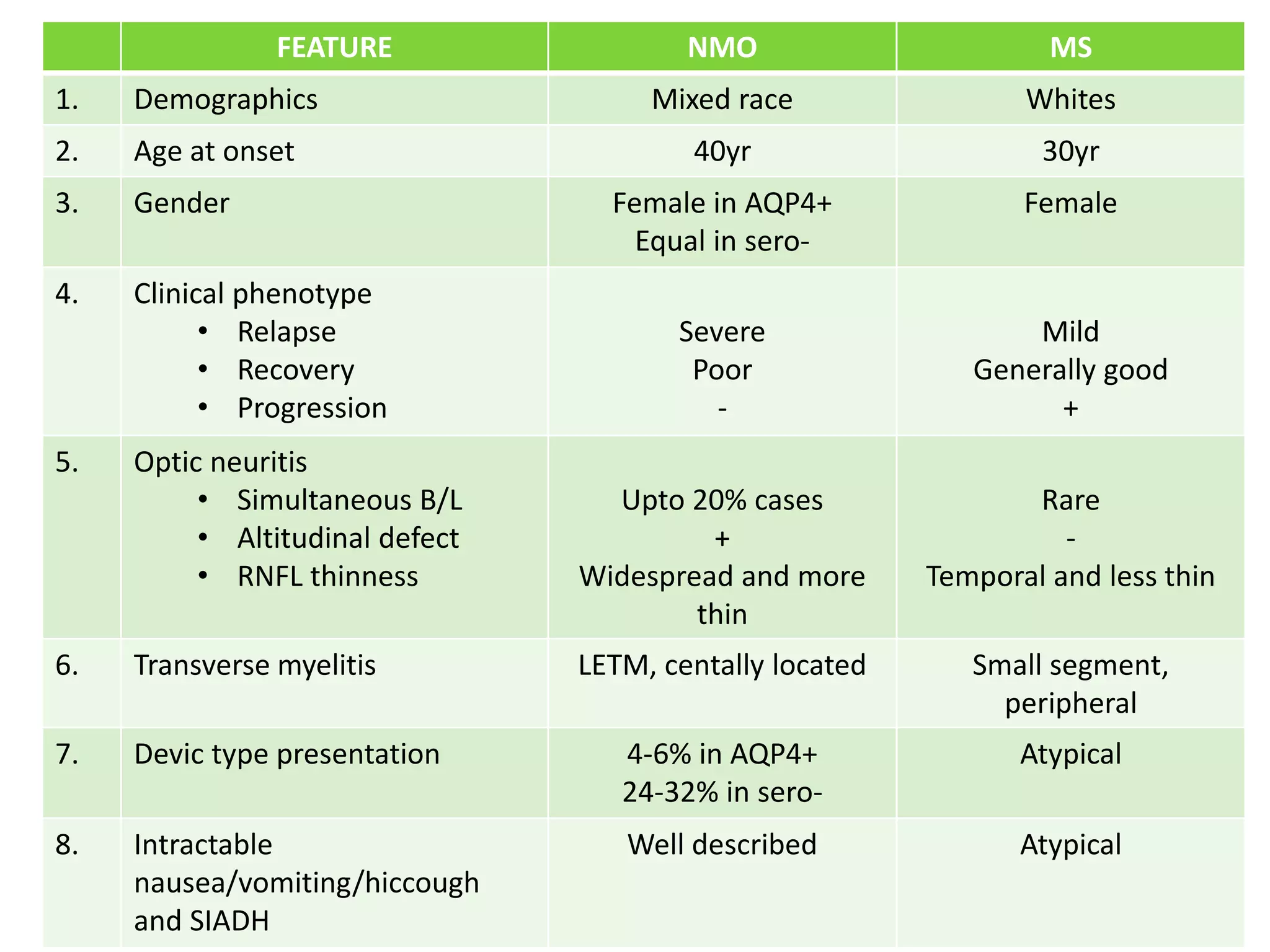
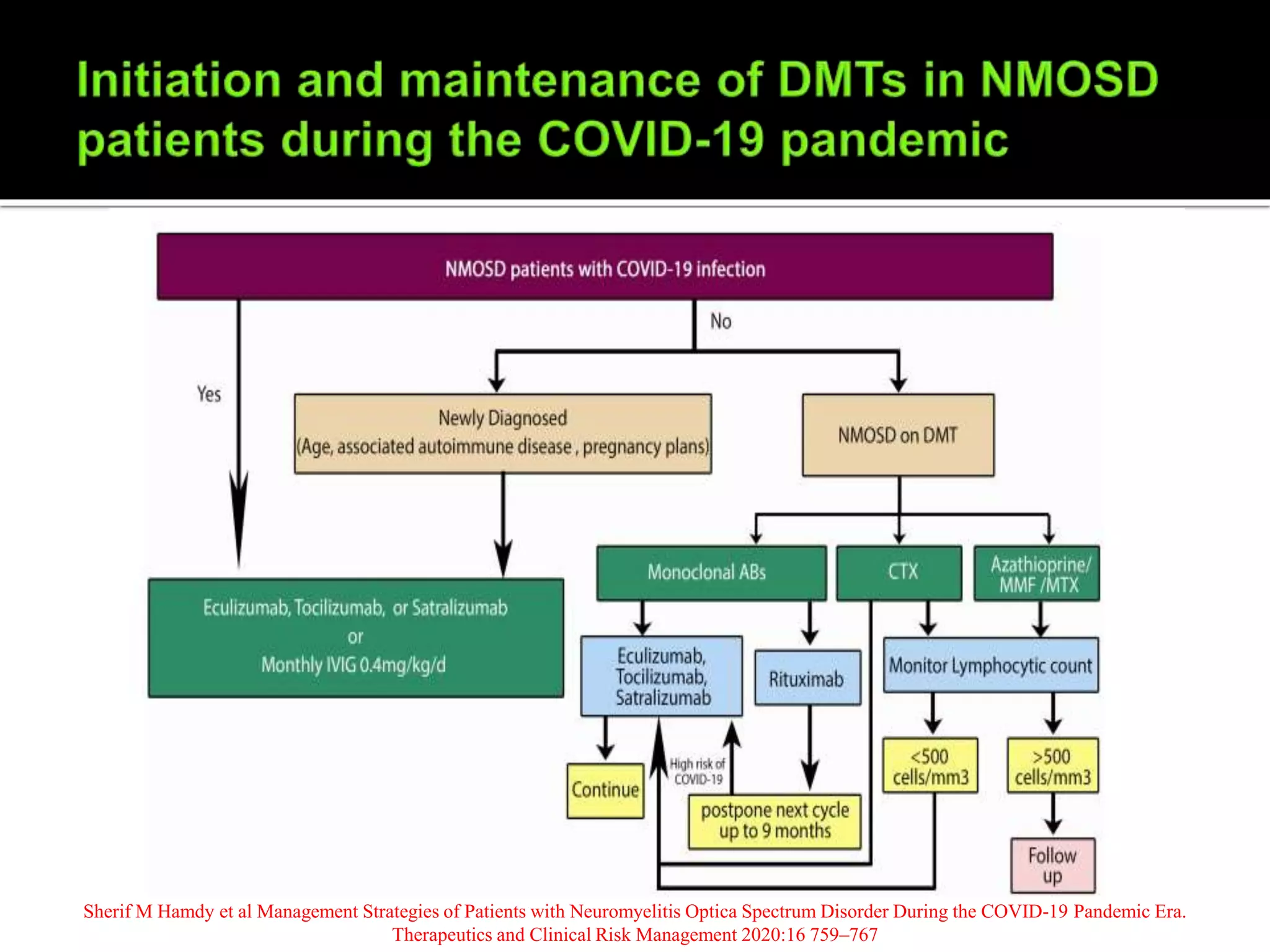
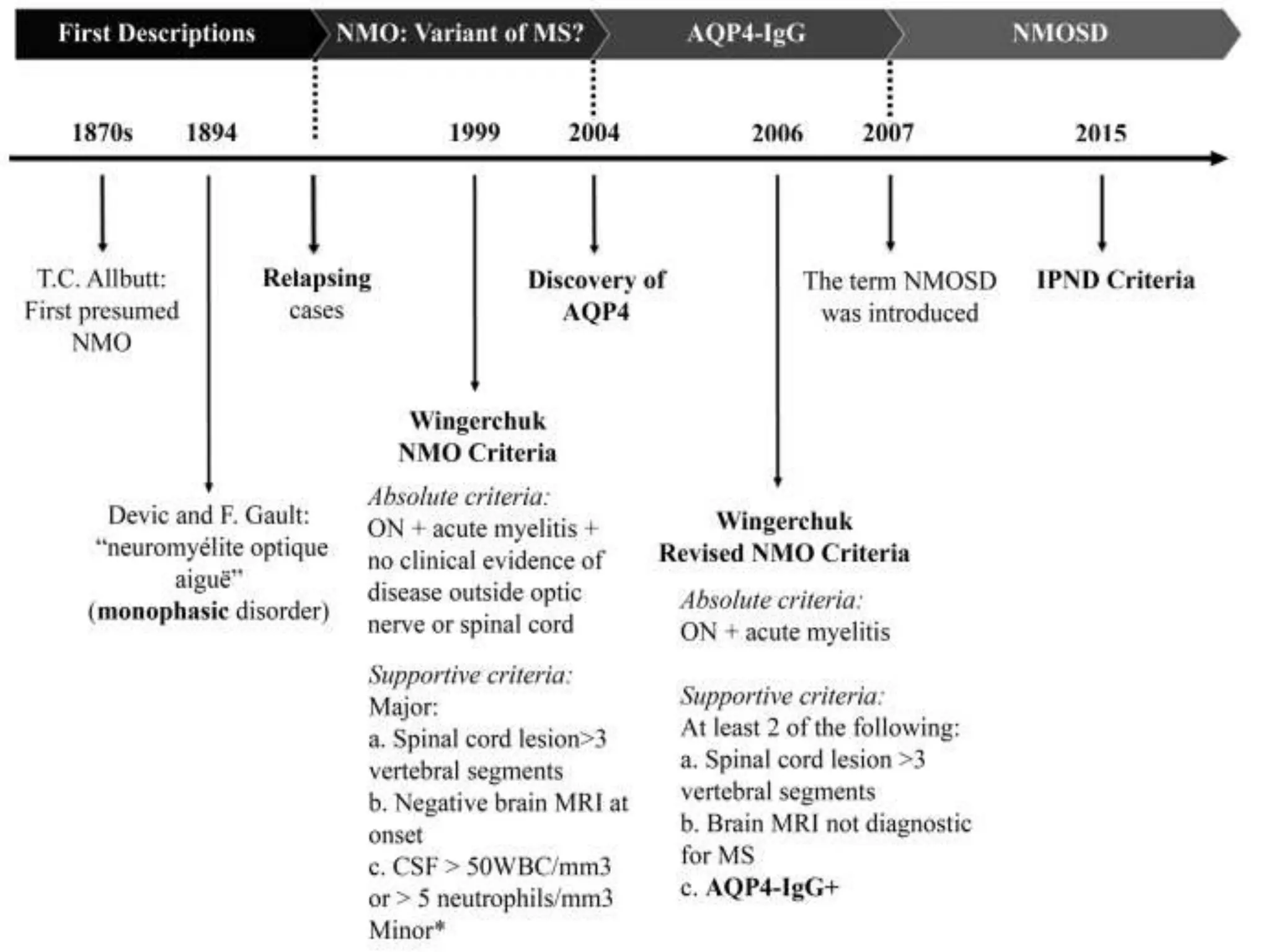
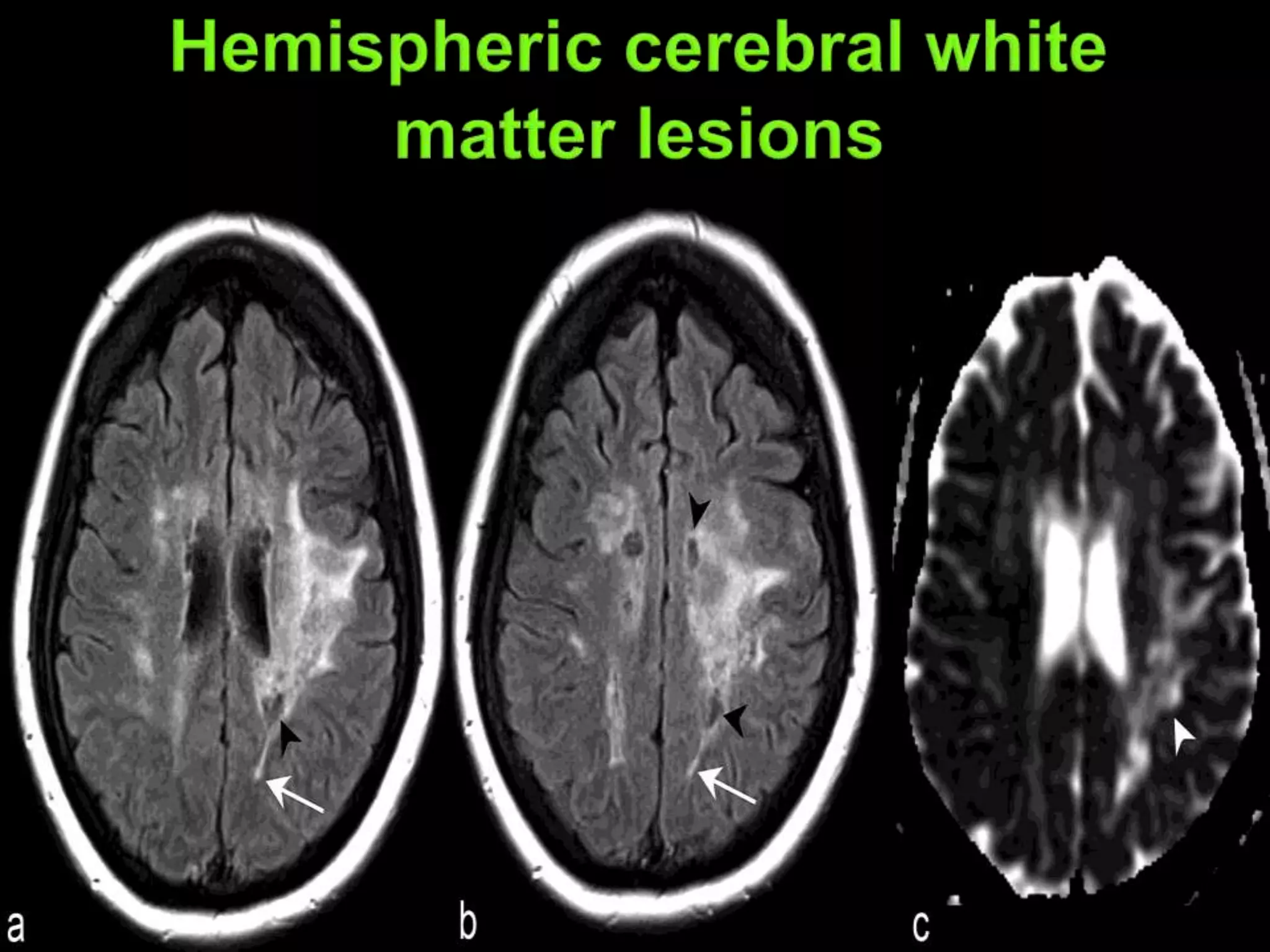
Dr. Shubham Garg discusses neuromyelitis optica (NMO), an autoimmune condition where antibodies attack aquaporin-4 in the central nervous system. NMO predominantly affects women and has a median age of onset of 32-41 years. Key clinical features include transverse myelitis, typically longitudinally extensive, and severe optic neuritis. Treatment involves high-dose steroids for acute attacks and immunosuppressants like azathioprine to reduce relapse rates. Prognosis is generally worse than multiple sclerosis due to risk of cumulative disability, though relapse rates can be lowered with appropriate treatment.




![Cardinal Clinical Features
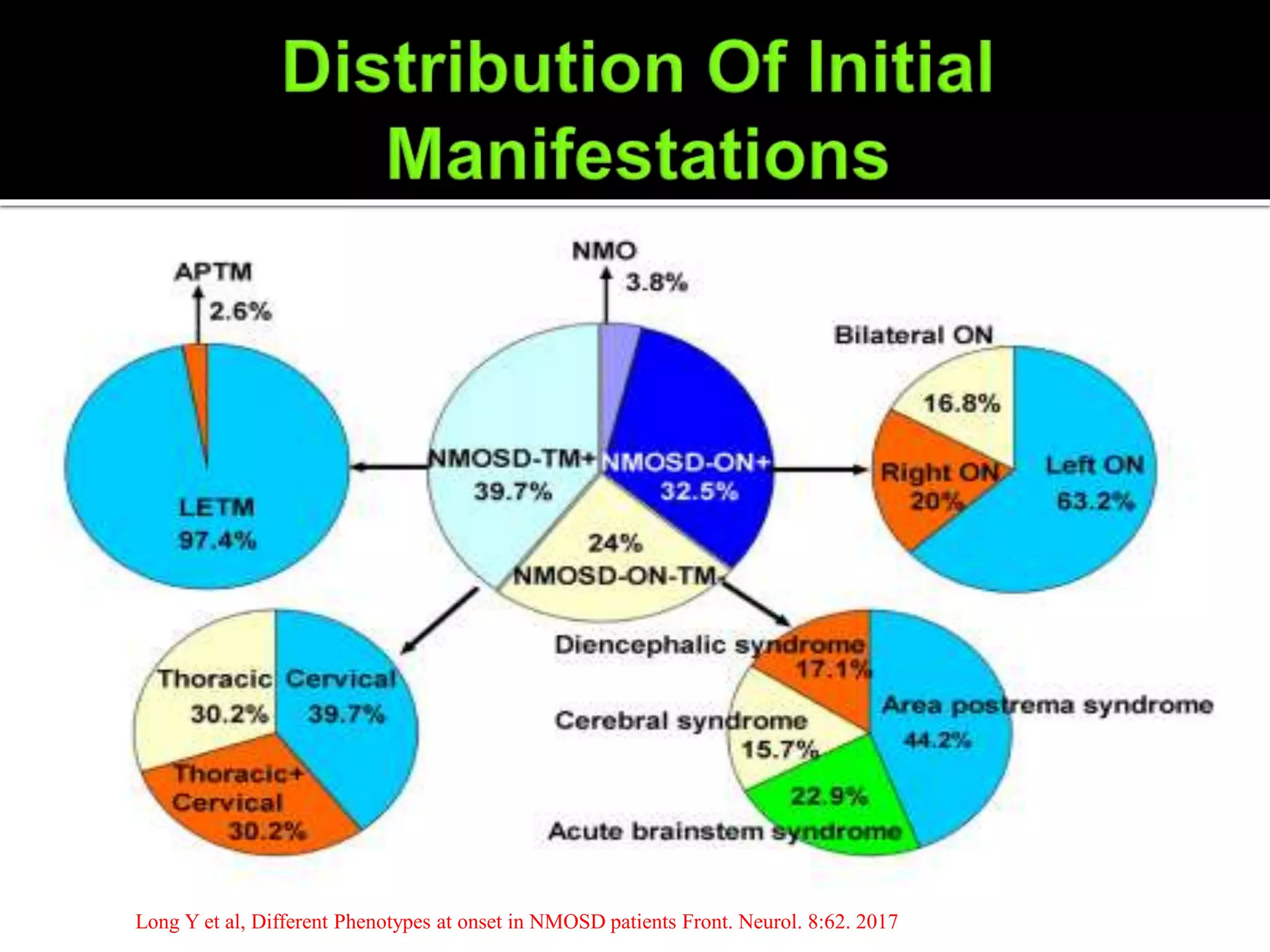
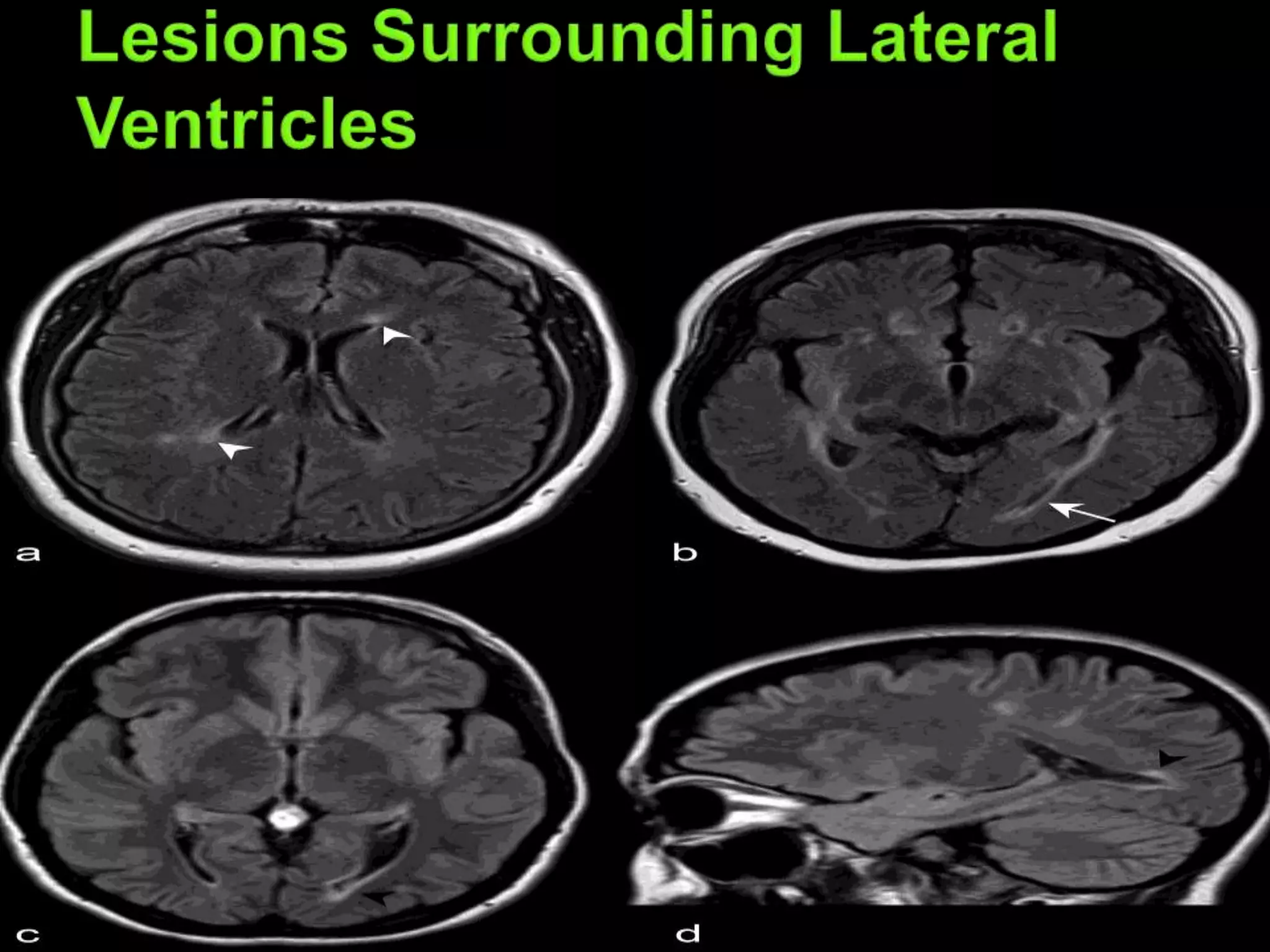
Transverse myelitis, typically longitudinally extensive (≥3 vertebral segments;
often followed by tonic spasms and occasionally accompanied by pain or pruritus)
Optic neuritis (often severe; may be bilateral)
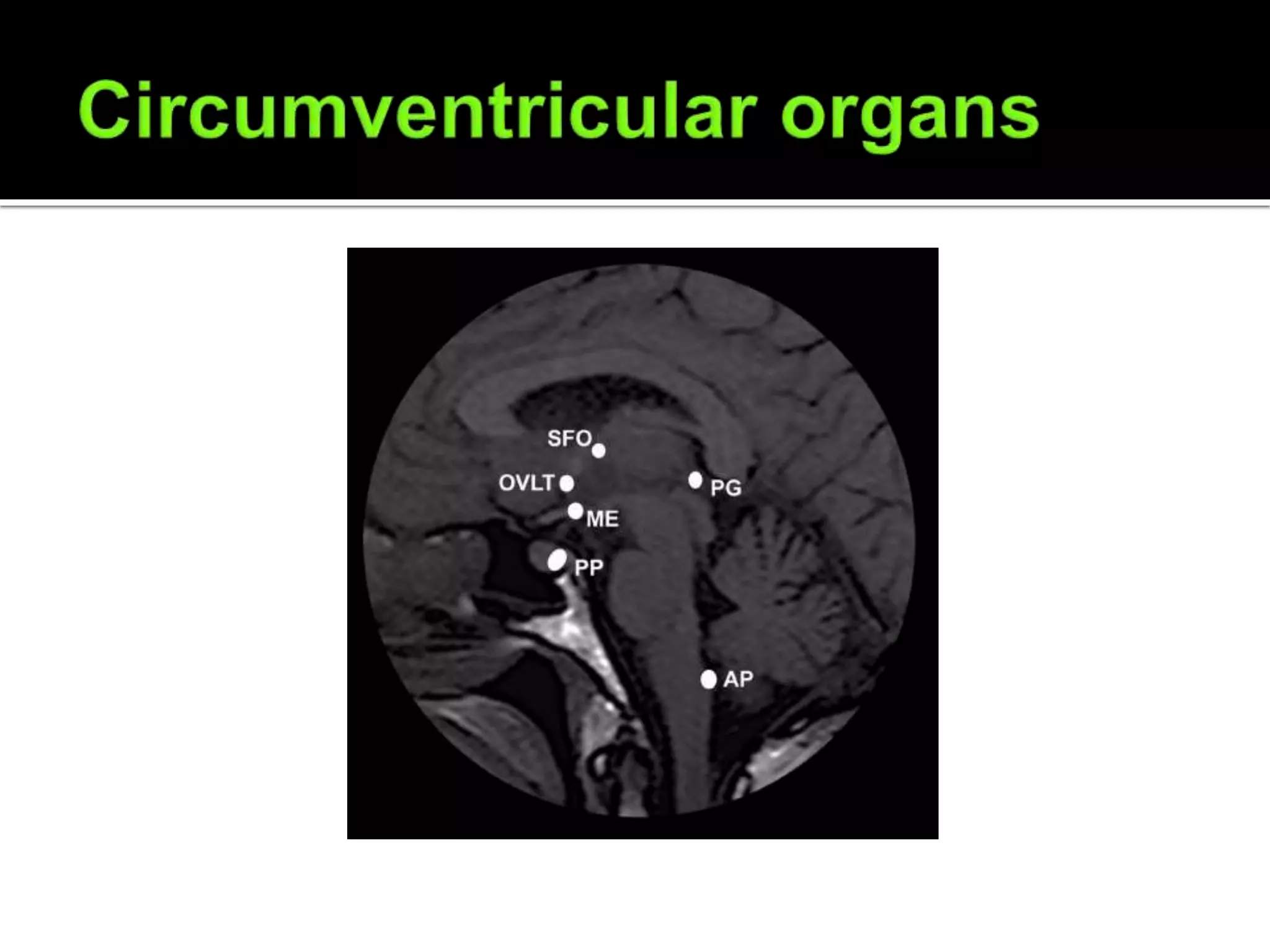
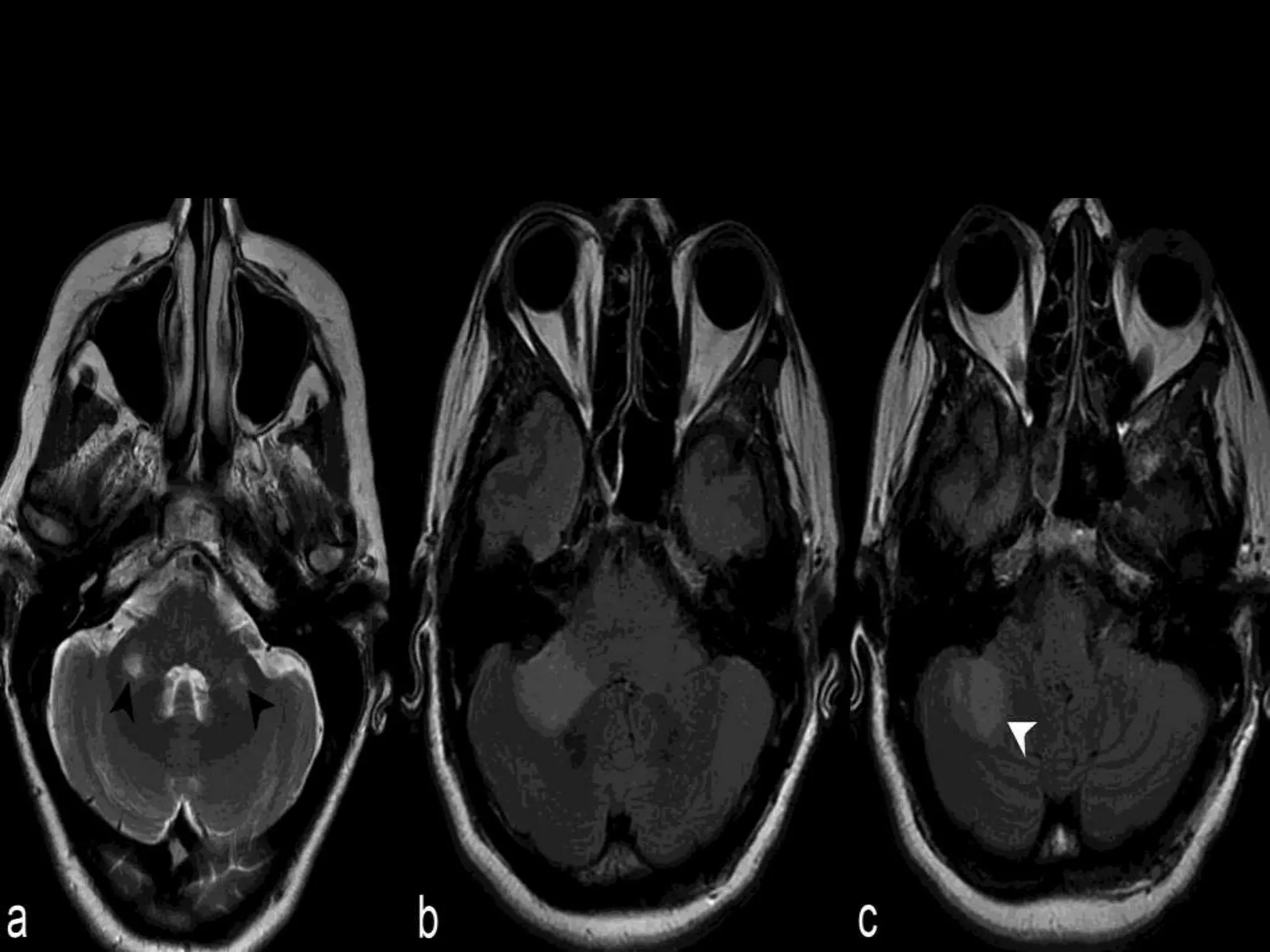
Episodes of intractable nausea and vomiting or hiccups from area postrema
involvement
Other Clinical Features
Narcolepsy
Syndrome of inappropriate secretion of antidiuretic hormone (SIADH)
Other hypothalamic presentations (eg, anorexia)
Acute myopathy with hyperCKemia
Brainstem syndromes (eg, ophthalmoplegia, hearing loss [possibly related to inner
ear damage] opsoclonus/myoclonus)
Myeloradiculitis
Encephalopathy (PRES-like; ADEM-like)
Cognitive dysfunction (subcortical pattern [inattention, executive dysfunction,
reduced speed of processing])](https://image.slidesharecdn.com/nmosdmog-211024134104/75/Nmosd-mog-5-2048.jpg)





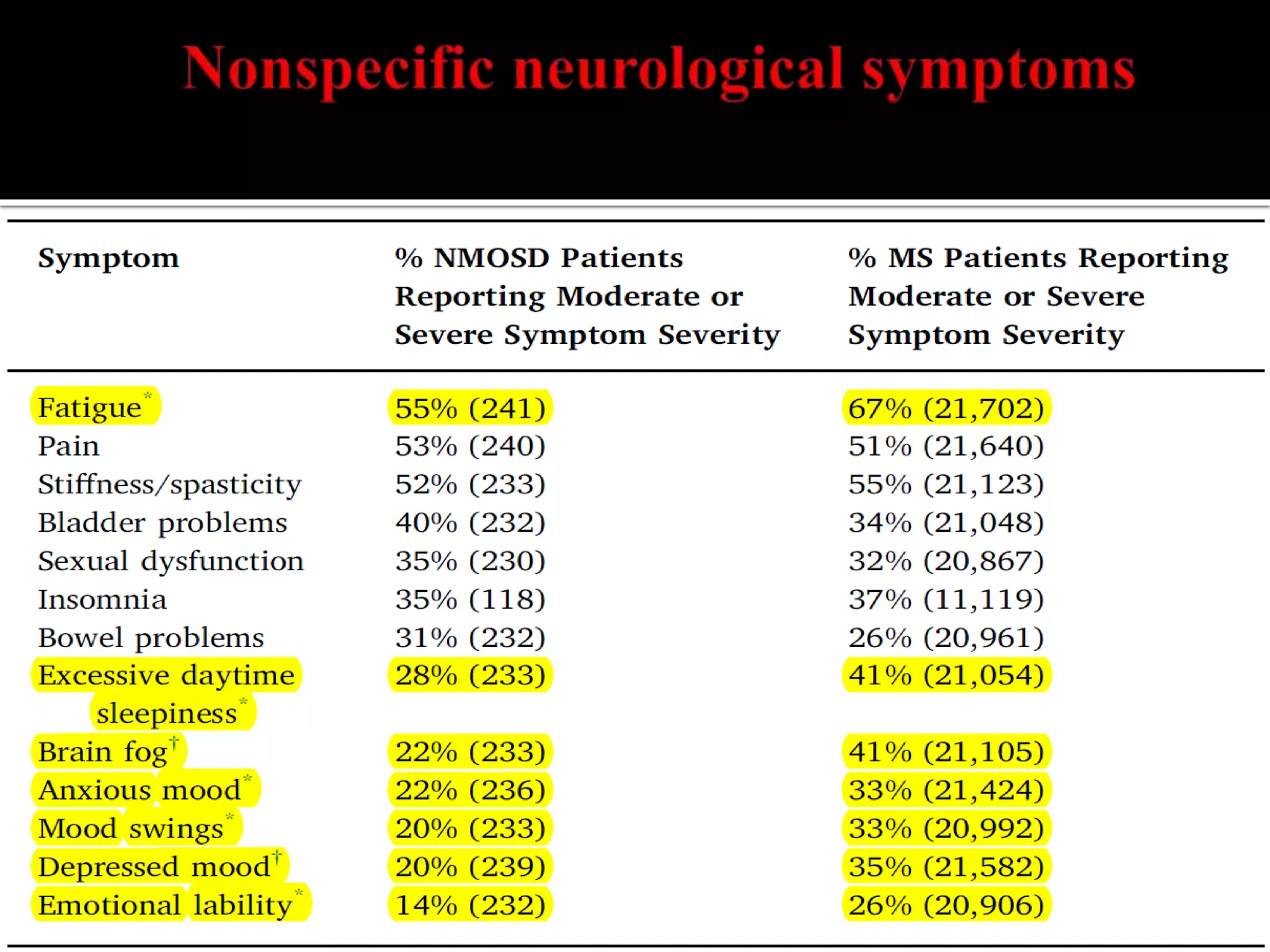
![ Sudden-onset dystonic posturing either uni- or bilaterally with
a stereotyped pattern
Brief, lasting less than 2 min, several times/hour
Incidence higher in the patients with NMO (10 patients
[25.0%]) than in those with multiple sclerosis (1 patient
[2.9%].
Painful tonic spasm associated with myelitis had a specificity
of 98.7% for identifying the NMO group
Ostermann PO, Westerberg CE.Brain.1975;98(2):189-202.](https://image.slidesharecdn.com/nmosdmog-211024134104/75/Nmosd-mog-12-2048.jpg)











































































![Myelin Oligodendrocyte Glycoprotein (MOG) Antibody Disease [MOG-AD]](https://cdn.slidesharecdn.com/ss_thumbnails/myelinoligodendrocyteglycoproteinmogantibodydisease0920-200920055159-thumbnail.jpg?width=640&height=640&fit=bounds)














































![DUAL AND TRIPLE ANTITHROMBOTIC THERAPY FOR SECONDARY STROKE [Autosaved].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/dualandtripleantithrombotictherapyforsecondarystrokeautosaved-230904113552-c3502b37-thumbnail.jpg?width=640&height=640&fit=bounds)


































