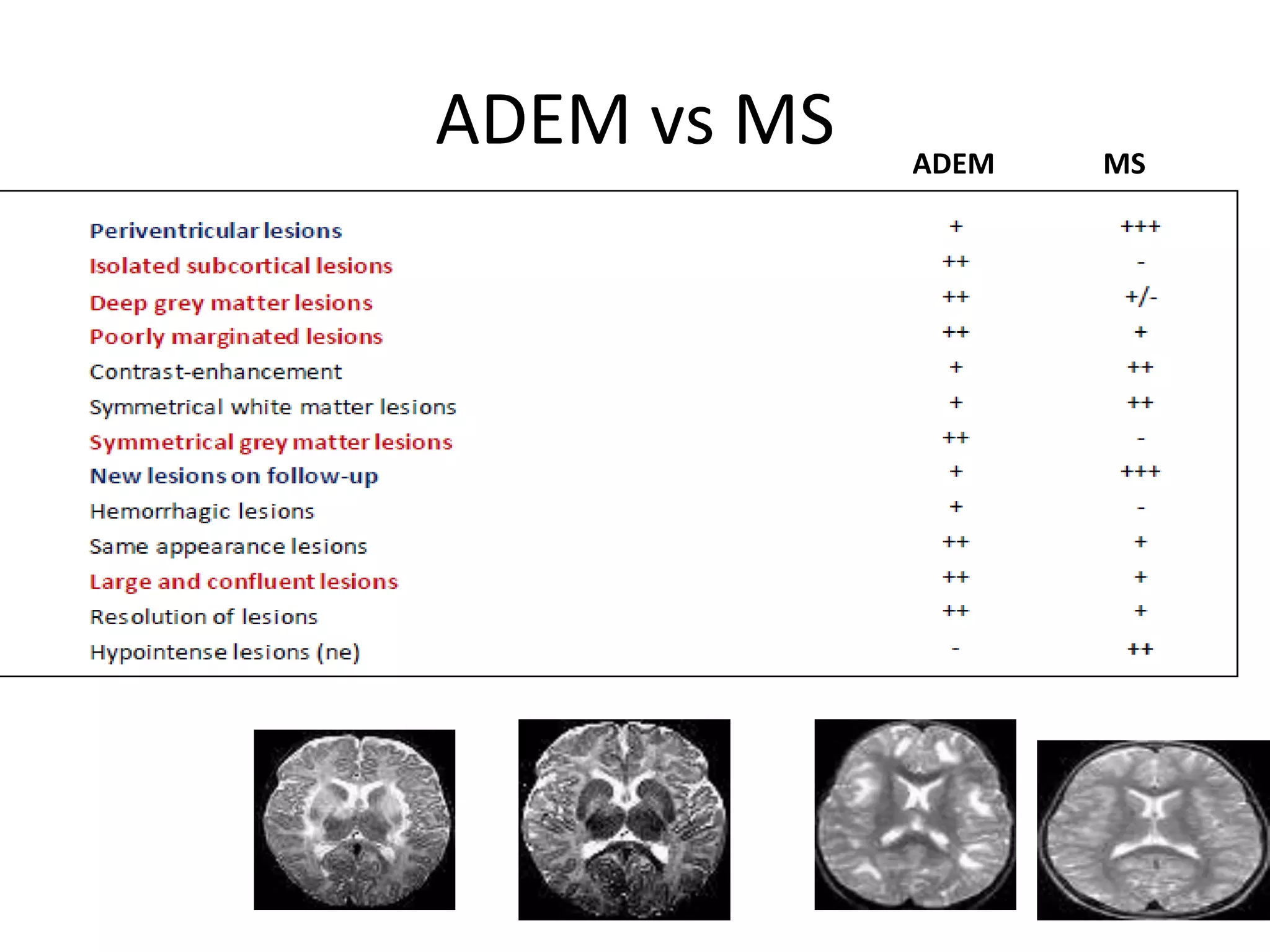
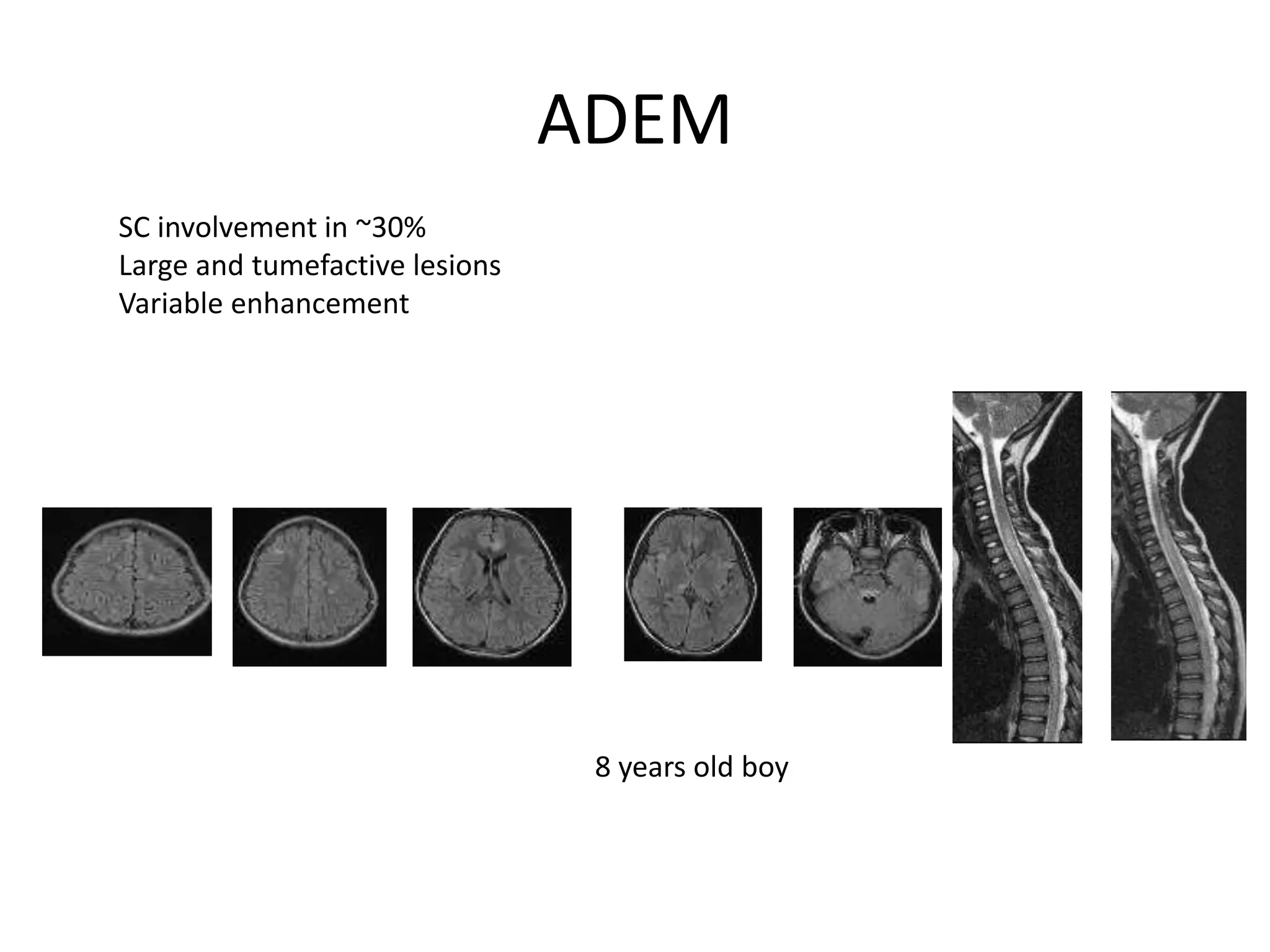
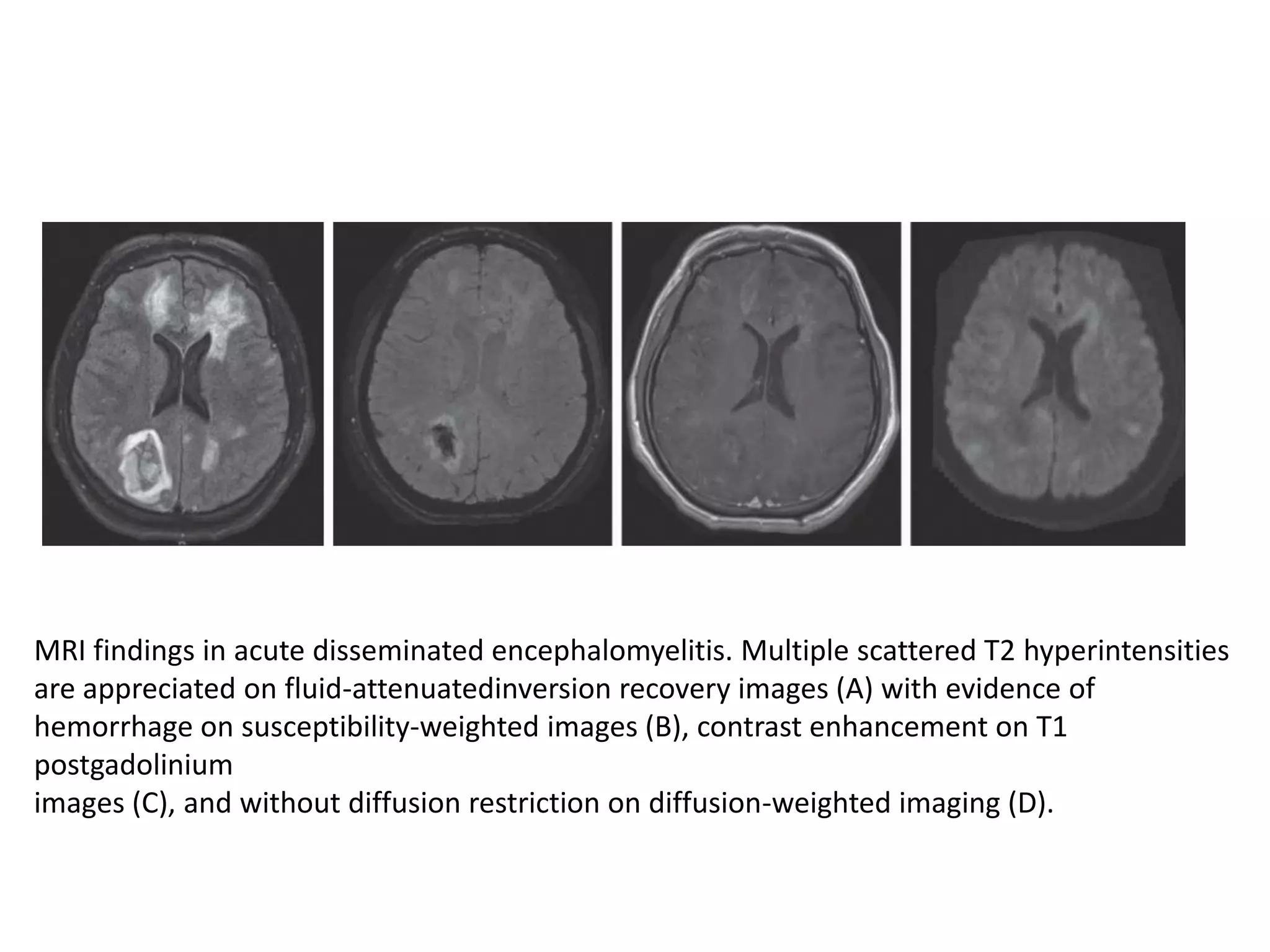
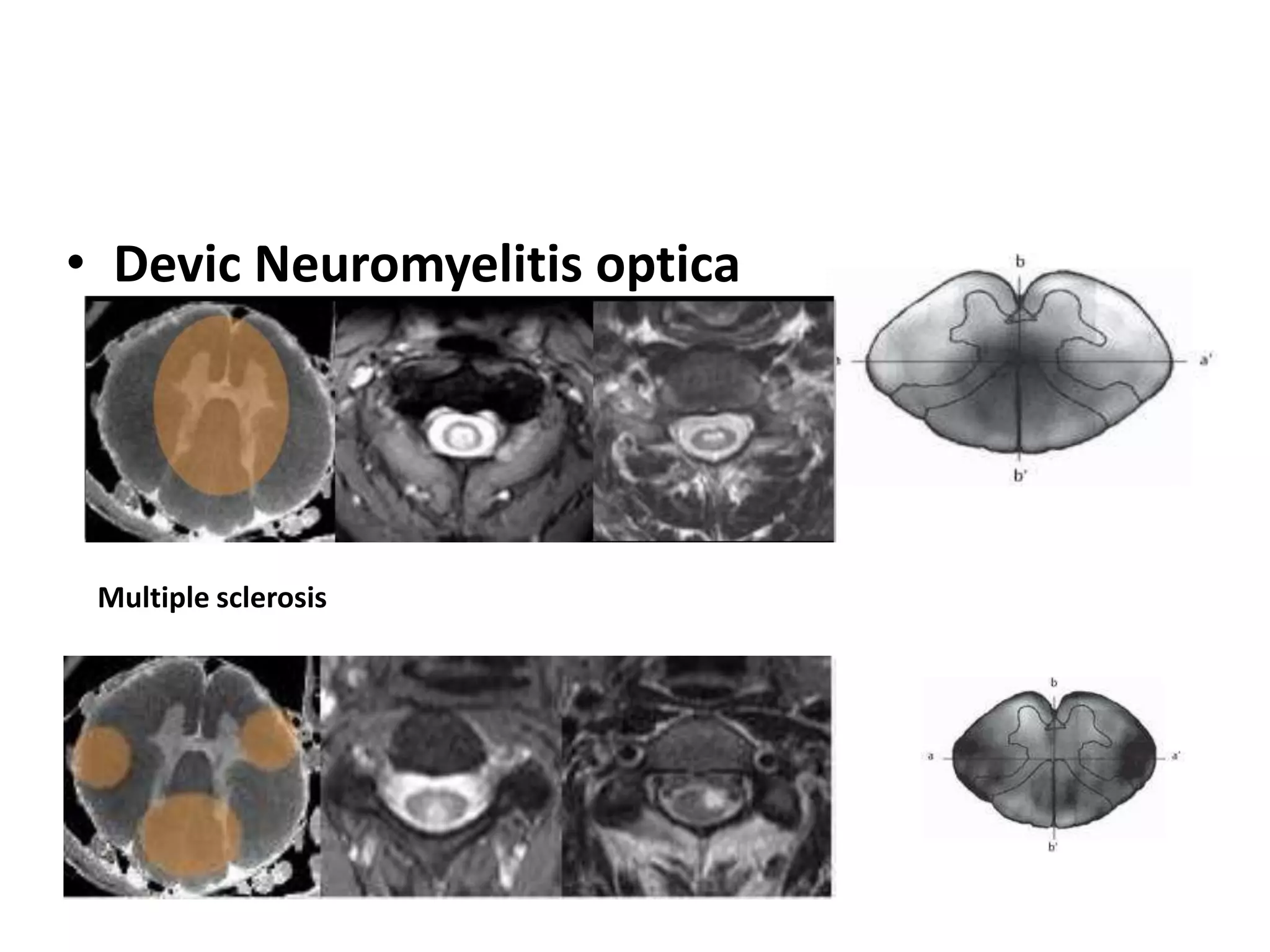
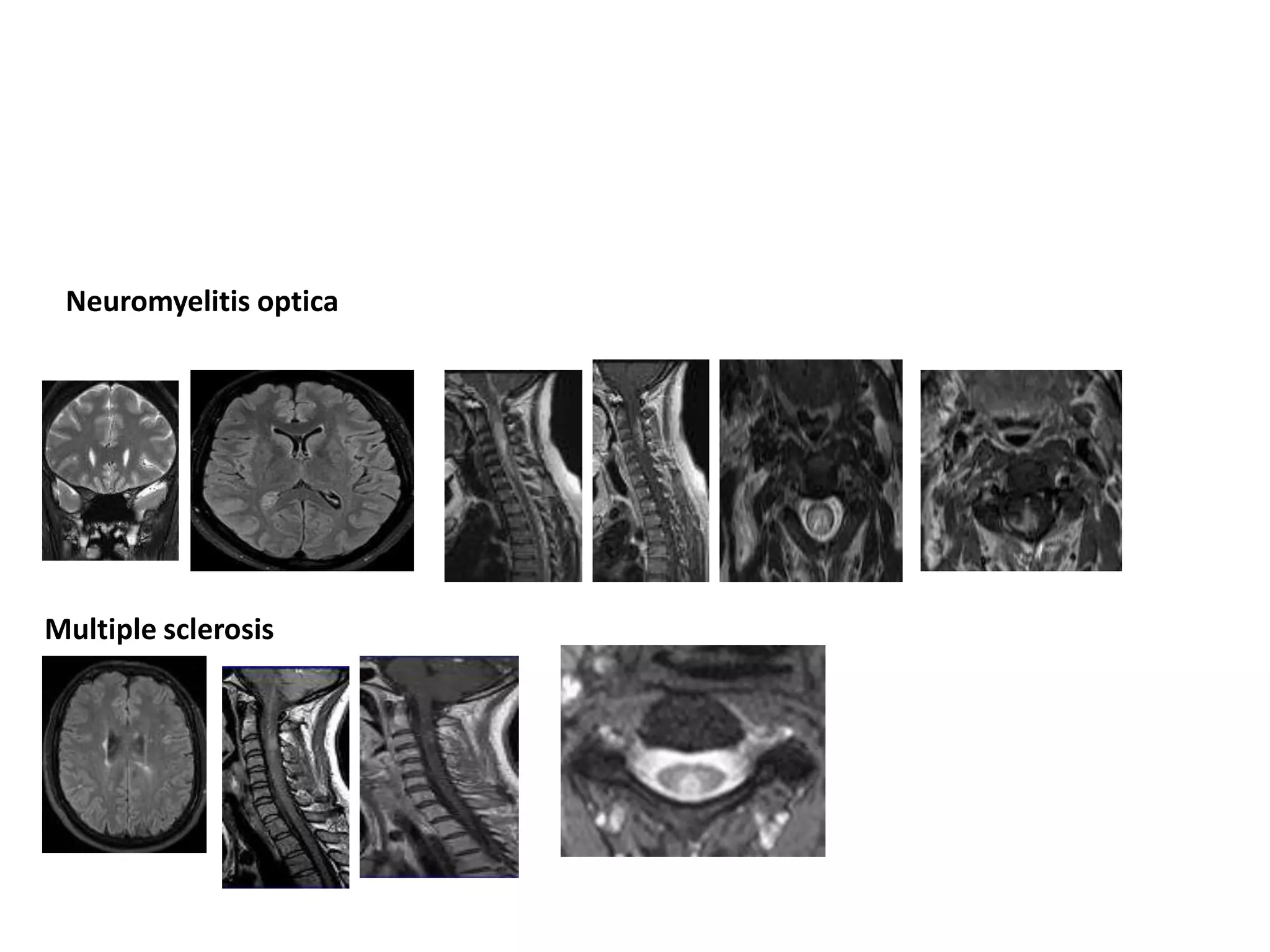
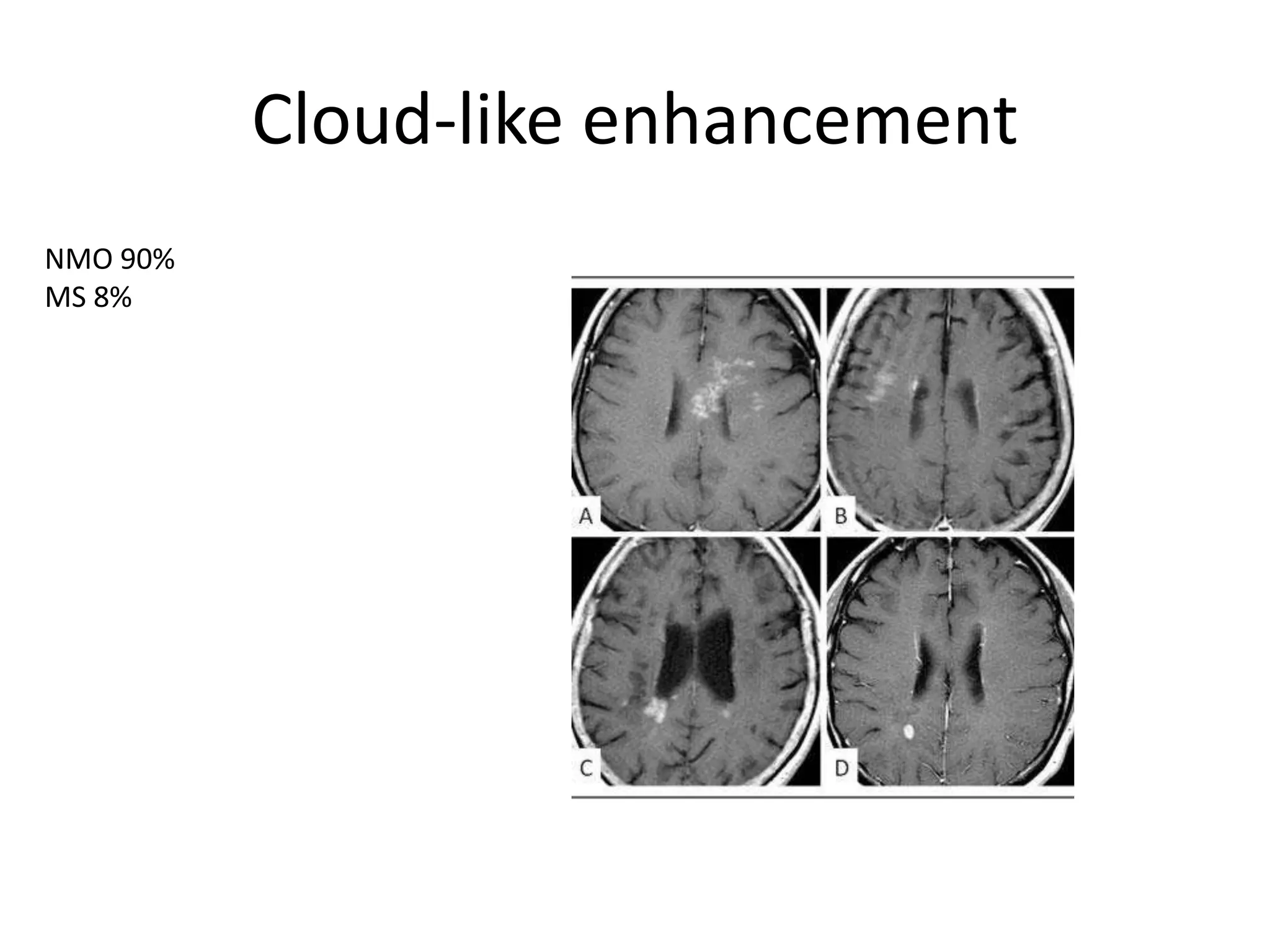
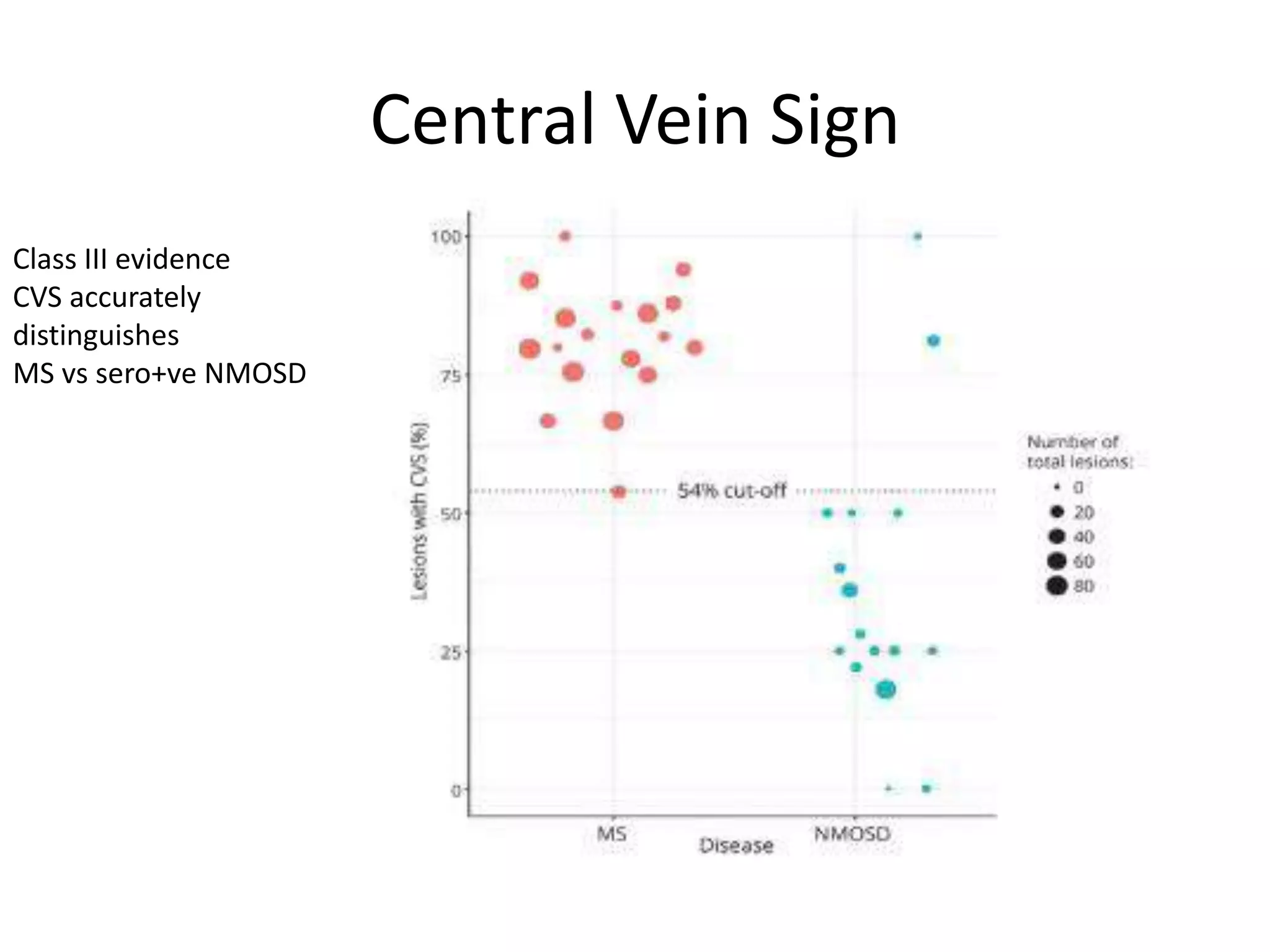
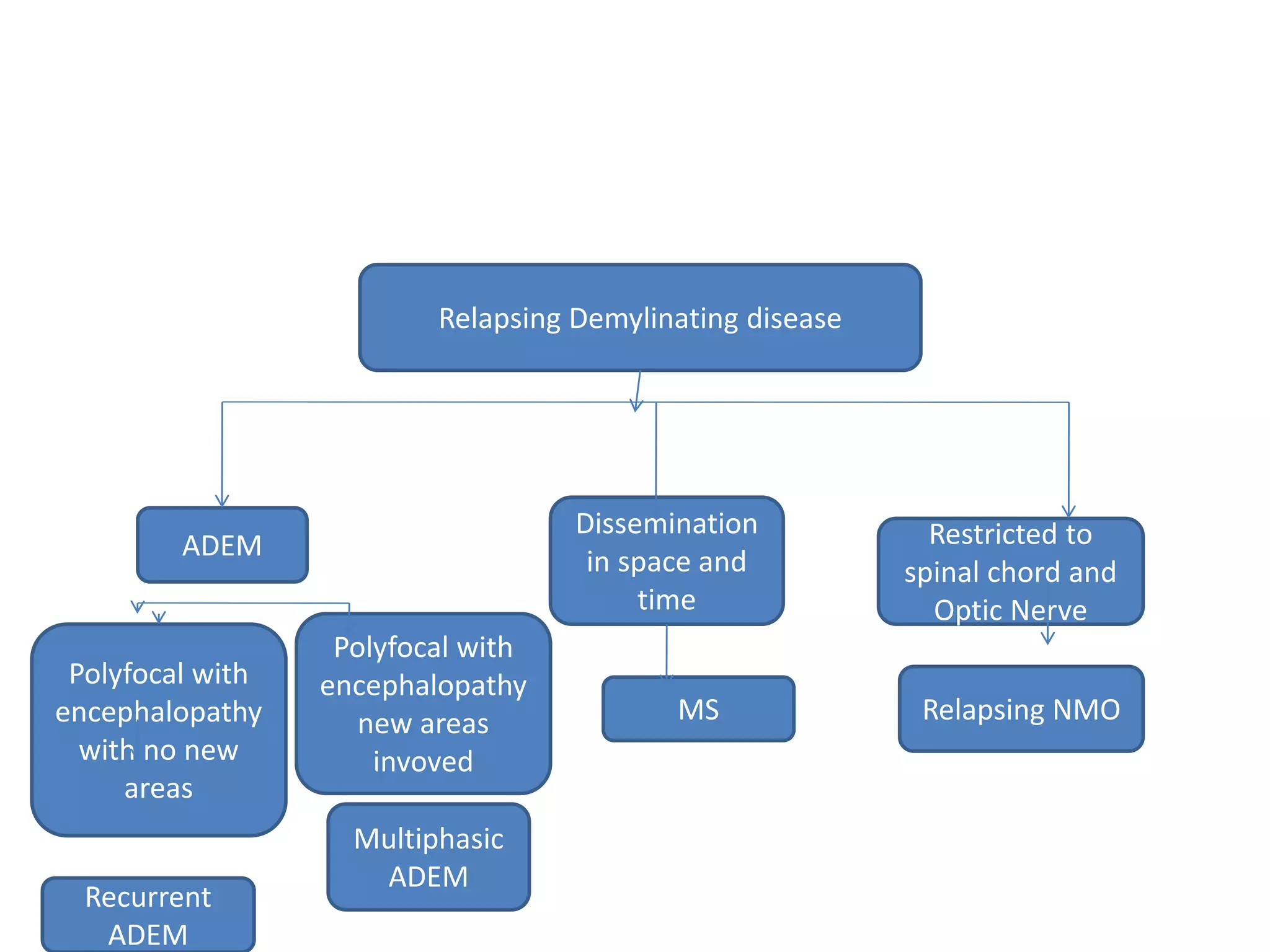
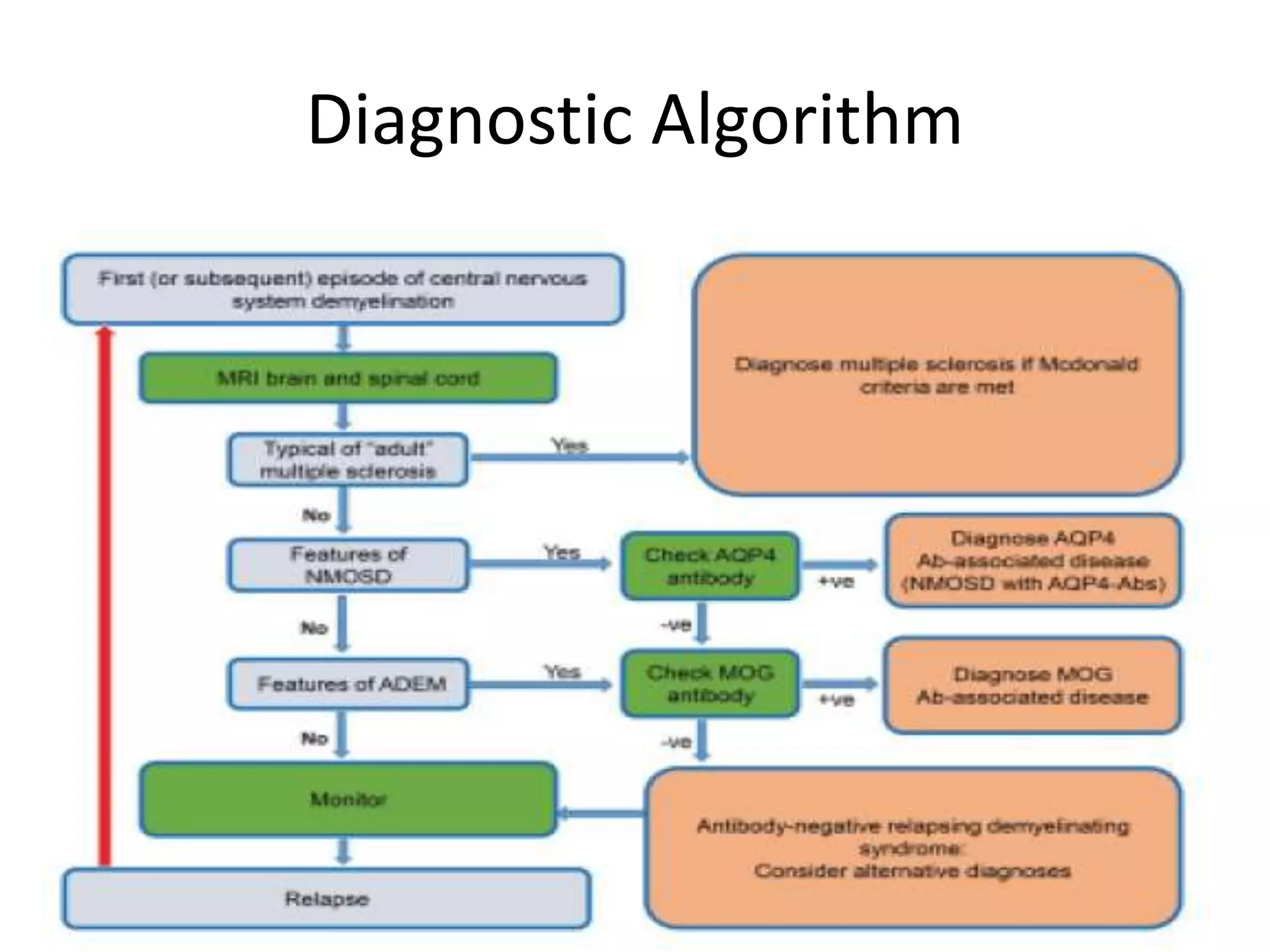
The document discusses demyelinating diseases, highlighting the clinical features and diagnostic criteria for conditions such as multiple sclerosis and acute disseminated encephalomyelitis (ADEM). Key indicators for suspecting demyelination include unexplained neurologic deficits, acute onset, and characteristic symptoms associated with specific demyelinating disorders. It also covers the evolution of different demyelinating conditions, their causes, and includes a case study illustrating the diagnostic approach to a patient with a possible demyelinating disease.






![Demylinating Disorders
• Immune mediated attack on white matter
including brain, optic nerve or spinal chord
• characterized clinically by
• localization of neurologic deficits (i.e., single site, such as spinal
cord [transverse myelitis], optic nerves [optic neuritis] or brainstem,
vs polyregional demyelination);
• the presence vs absence of encephalopathy; and
• disease course (i.e., monophasic vs repeated attacks involving
either the same or new CNS regions).](https://image.slidesharecdn.com/approachtodemyelinatingdiseases-210914044202/75/Approach-to-demyelinating-diseases-7-2048.jpg)






















































































![DUAL AND TRIPLE ANTITHROMBOTIC THERAPY FOR SECONDARY STROKE [Autosaved].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/dualandtripleantithrombotictherapyforsecondarystrokeautosaved-230904113552-c3502b37-thumbnail.jpg?width=640&height=640&fit=bounds)

































