Downloaded 93 times











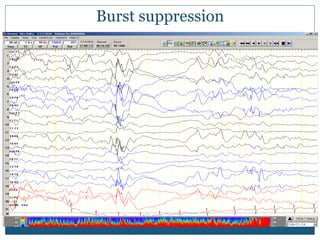



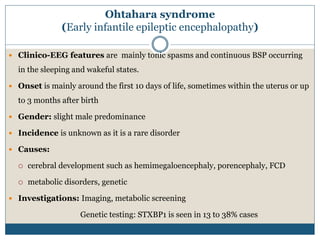





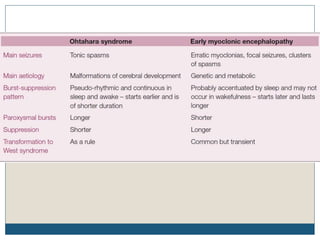






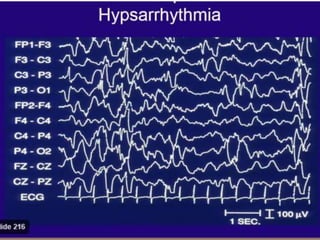
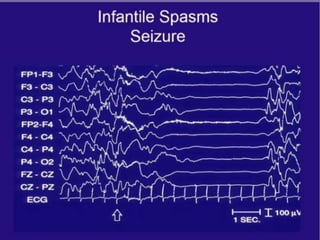
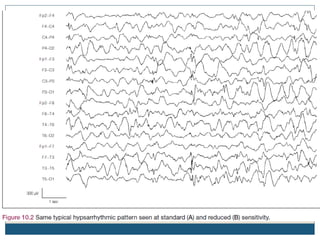











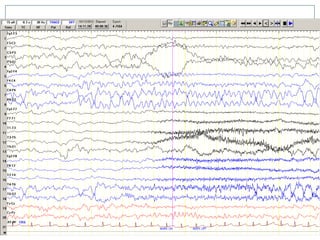








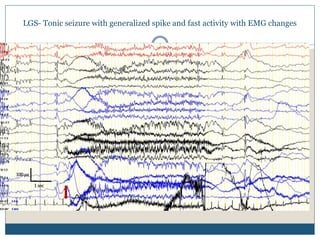
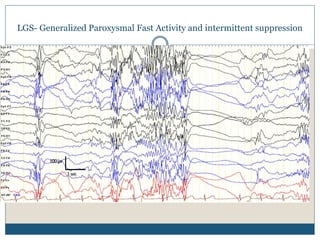
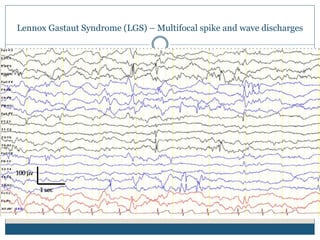
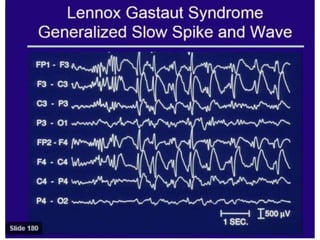
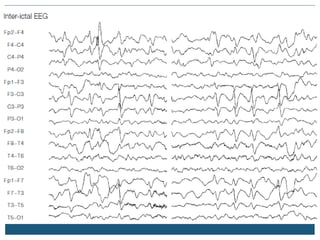




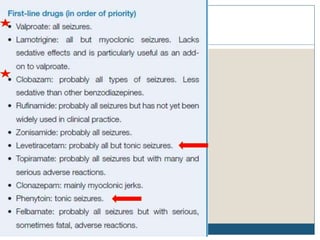
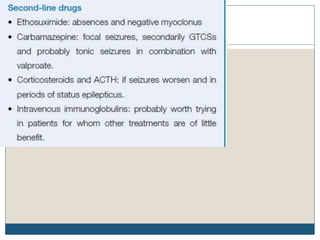





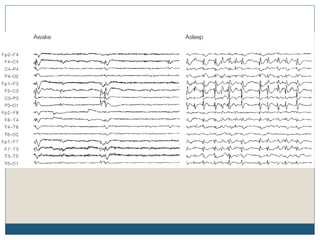
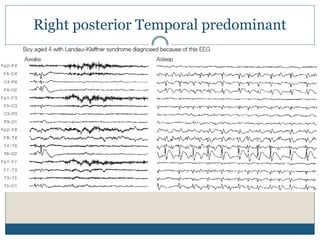




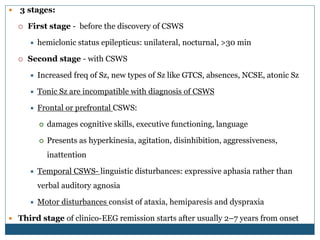

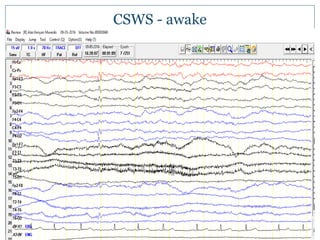
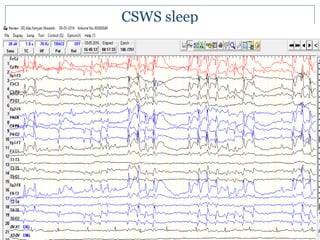
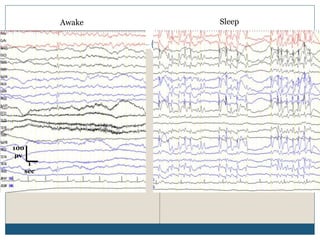


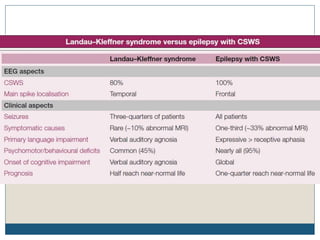





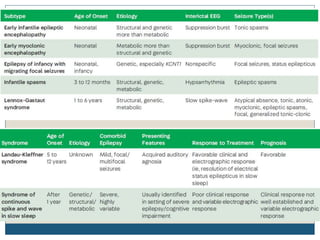

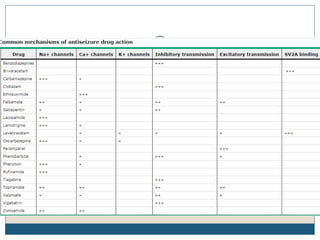


The document discusses various epileptic encephalopathies that typically begin early in life and are characterized by seizures, abnormal EEG patterns, and cognitive and neurological deterioration. It defines epileptic encephalopathies and provides details on specific syndromes including early myoclonic encephalopathy, Ohtahara syndrome, West syndrome, Dravet syndrome, and Lennox-Gastaut syndrome. For each syndrome, it discusses age of onset, causes, clinical features, EEG findings, treatment approaches, and prognosis.























































































