Downloaded 293 times















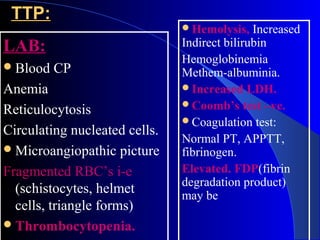

















This document discusses various bleeding disorders including definitions, etiologies, and clinical features. It covers vessel wall abnormalities like hereditary hemorrhagic telangiectasia and Ehlers Danlos disease. Platelet disorders discussed include quantitative issues like thrombocytopenia and qualitative issues such as Bernard Soulier syndrome. Coagulation disorders covered include hemophilia A, hemophilia B, and acquired disorders like disseminated intravascular coagulation and liver disease. Specific conditions like idiopathic thrombocytopenic purpura, thrombotic thrombocytopenic purpura, and von Willebrand disease are explained in detail.








































![[Int. med] bleeding disorders from SIMS Lahore](https://cdn.slidesharecdn.com/ss_thumbnails/4me9f7ogtuypxn18imbp-signature-b01672da1ecf8b94befb115319b147a085de390b8cb403389bce6c156545fbb5-poli-150815171702-lva1-app6891-thumbnail.jpg?width=640&height=640&fit=bounds)




























































