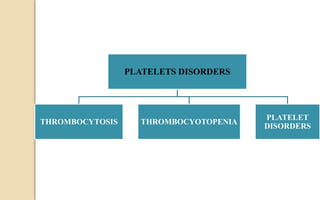
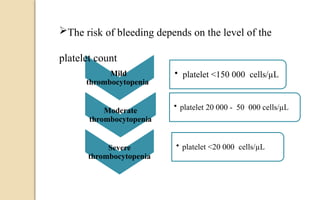
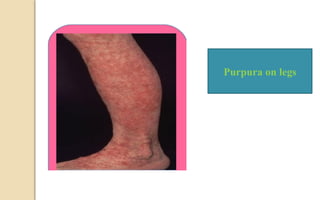
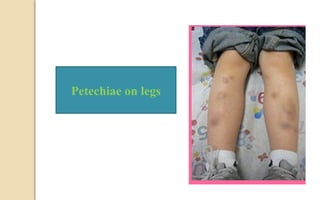
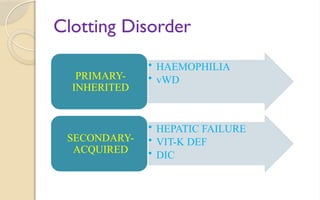
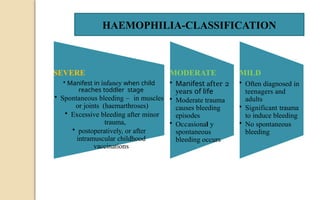
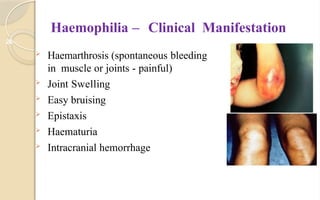
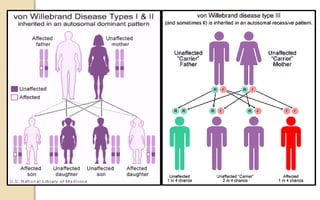
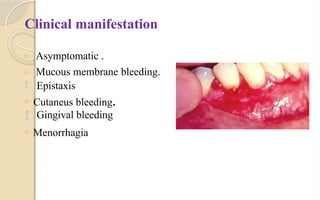
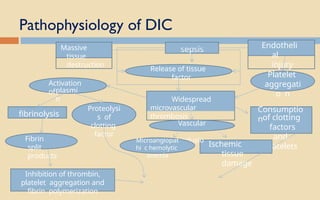
The document discusses platelet disorders, including thrombocytopenia and thrombocytosis, outlining their definitions, etiologies, clinical manifestations, and diagnostic approaches. It details the management and treatment options for these disorders, as well as implications for dental care, particularly in hemophilia and von Willebrand's disease. The text emphasizes the importance of a thorough history and clinical examination in diagnosing bleeding disorders.



























































![Hematology%20disorder%20in%20dental%20treatment[1]](https://cdn.slidesharecdn.com/ss_thumbnails/hematology20disorder20in20dental20treatment1-161105033248-thumbnail.jpg?width=640&height=640&fit=bounds)



























![Hypothalamus short ppt by Dr. Neha [PT].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/hypothalamusbydr-260124145759-b9f94a93-thumbnail.jpg?width=640&height=640&fit=bounds)