Downloaded 252 times








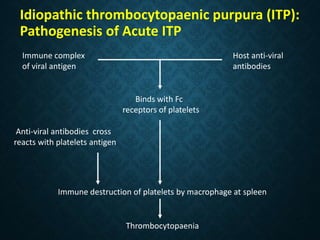
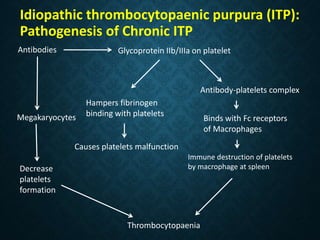


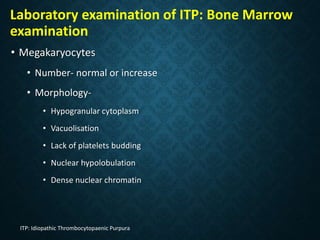

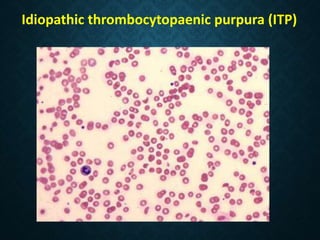


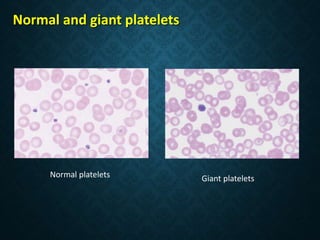

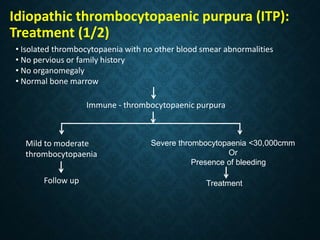

















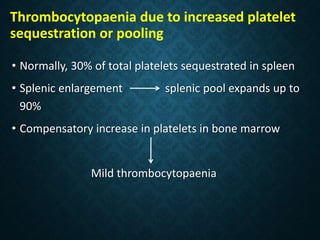

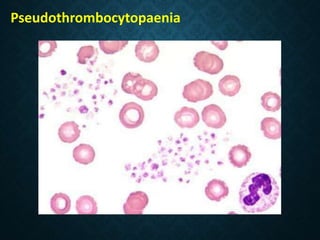



Thrombocytopaenia, or low platelet count, can be caused by decreased platelet production or increased platelet destruction. Causes of decreased production include bone marrow diseases and medications. Increased destruction can be due to immune-mediated causes like Idiopathic Thrombocytopenic Purpura (ITP) or non-immune causes like disseminated intravascular coagulation. ITP is caused by autoantibodies that bind to and destroy platelets, and presents with mild bleeding and a normal bone marrow with increased megakaryocytes. Thrombocytopaenia is diagnosed based on blood counts, smear, and ruling out other potential causes through testing and history. Treatment depends on severity but









































































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)

![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)





