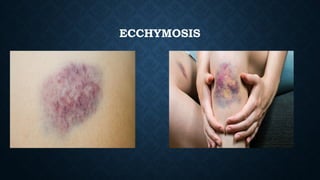
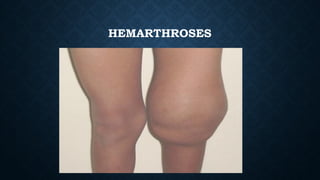
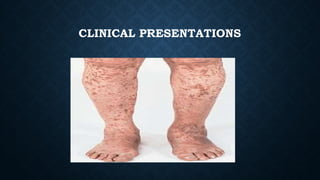
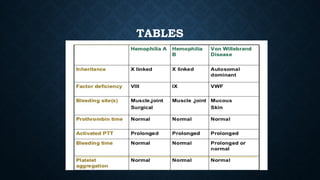
The document provides an overview of bleeding disorders, including haemophilia A and B, immune/idiopathic thrombocytopenic purpura (ITP), and von Willebrand disease, detailing their pathophysiology, clinical presentations, and management strategies. Haemophilia is characterized by deficiencies in specific coagulation factors leading to excessive bleeding, while ITP results from immune destruction of platelets. Von Willebrand disease is noted as the most common inherited bleeding disorder, featuring variable symptoms linked to von Willebrand factor dysfunction.










































































![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)







![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)
![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)





