Downloaded 24 times
























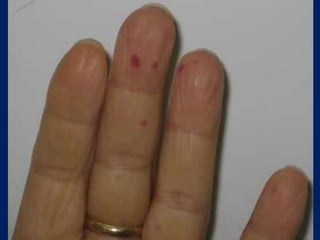


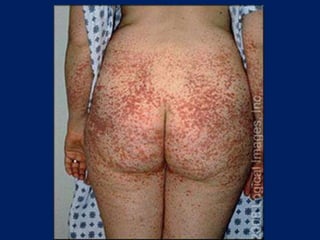

























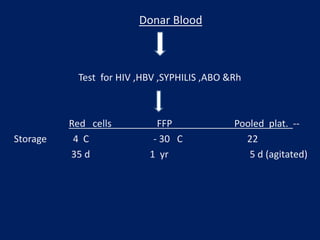
















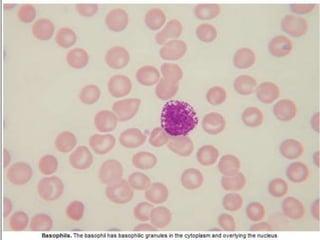





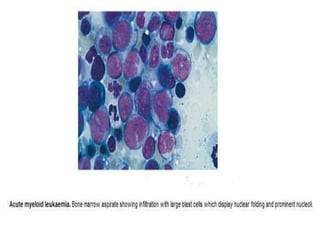

















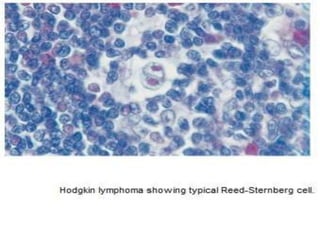
















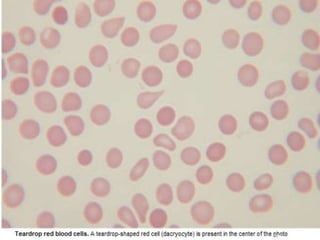






This document summarizes information on haemostasis and bleeding disorders. It discusses the mechanisms of haemostasis, screening tests for bleeding disorders, platelet disorders like ITP and TTP, coagulation factor deficiencies like hemophilia A and B, and vessel wall abnormalities. Key points covered are the stages of haemostasis, common causes of thrombocytopenia and thrombocytosis, management of ITP, and clinical features and treatment of disorders like hemophilia, von Willebrand disease, and Henoch-Schonlein purpura.