Downloaded 70 times








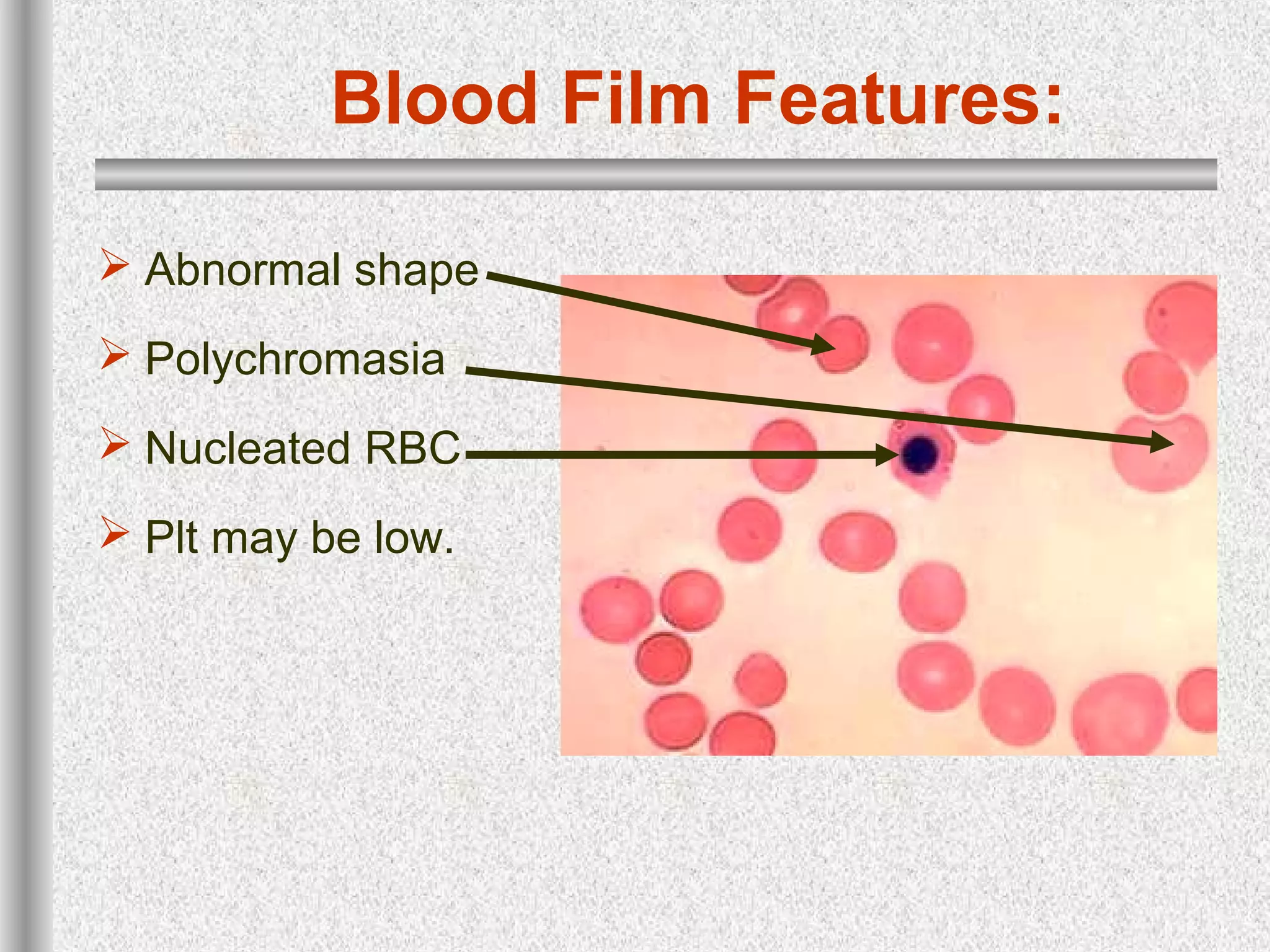







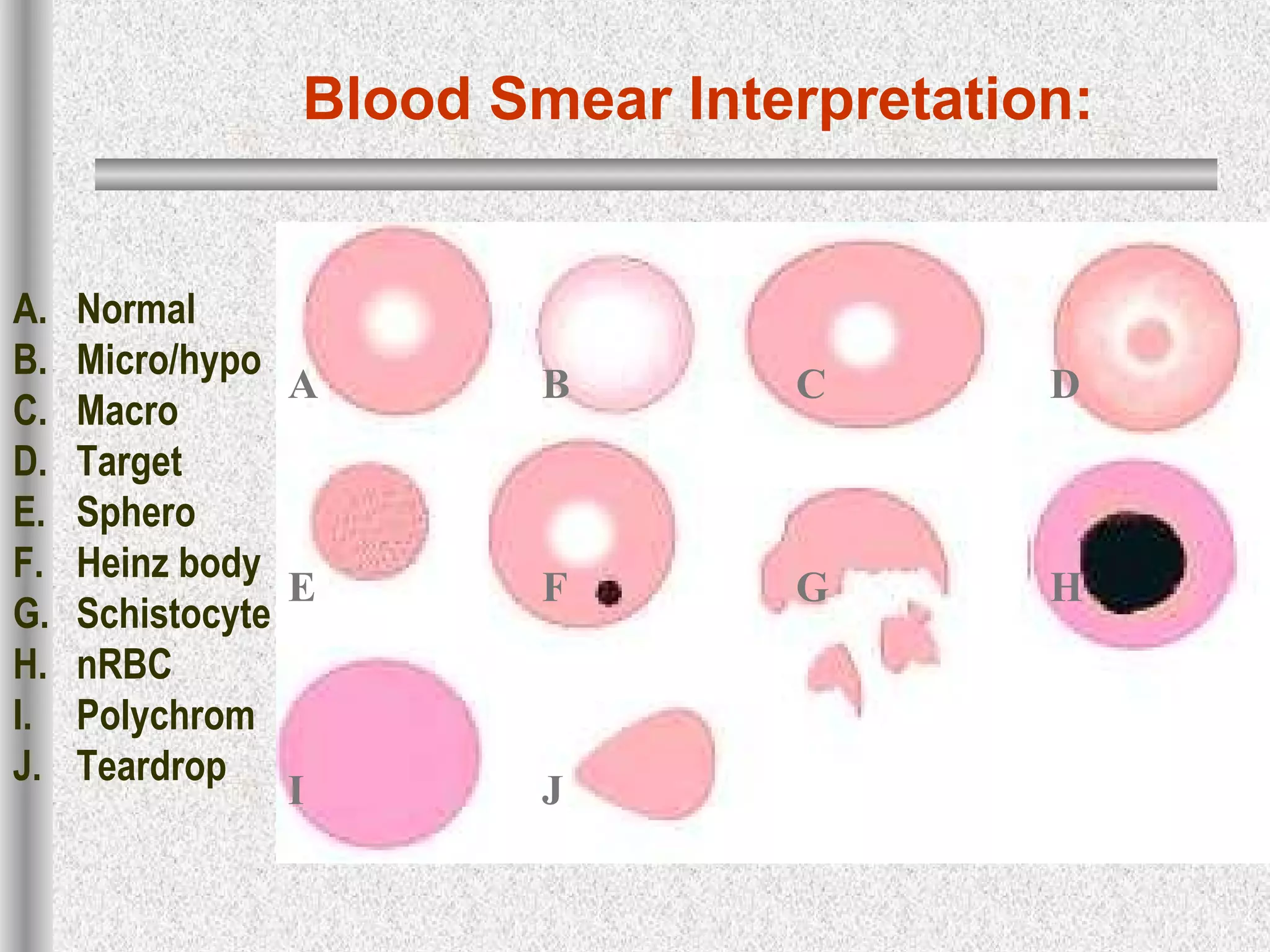



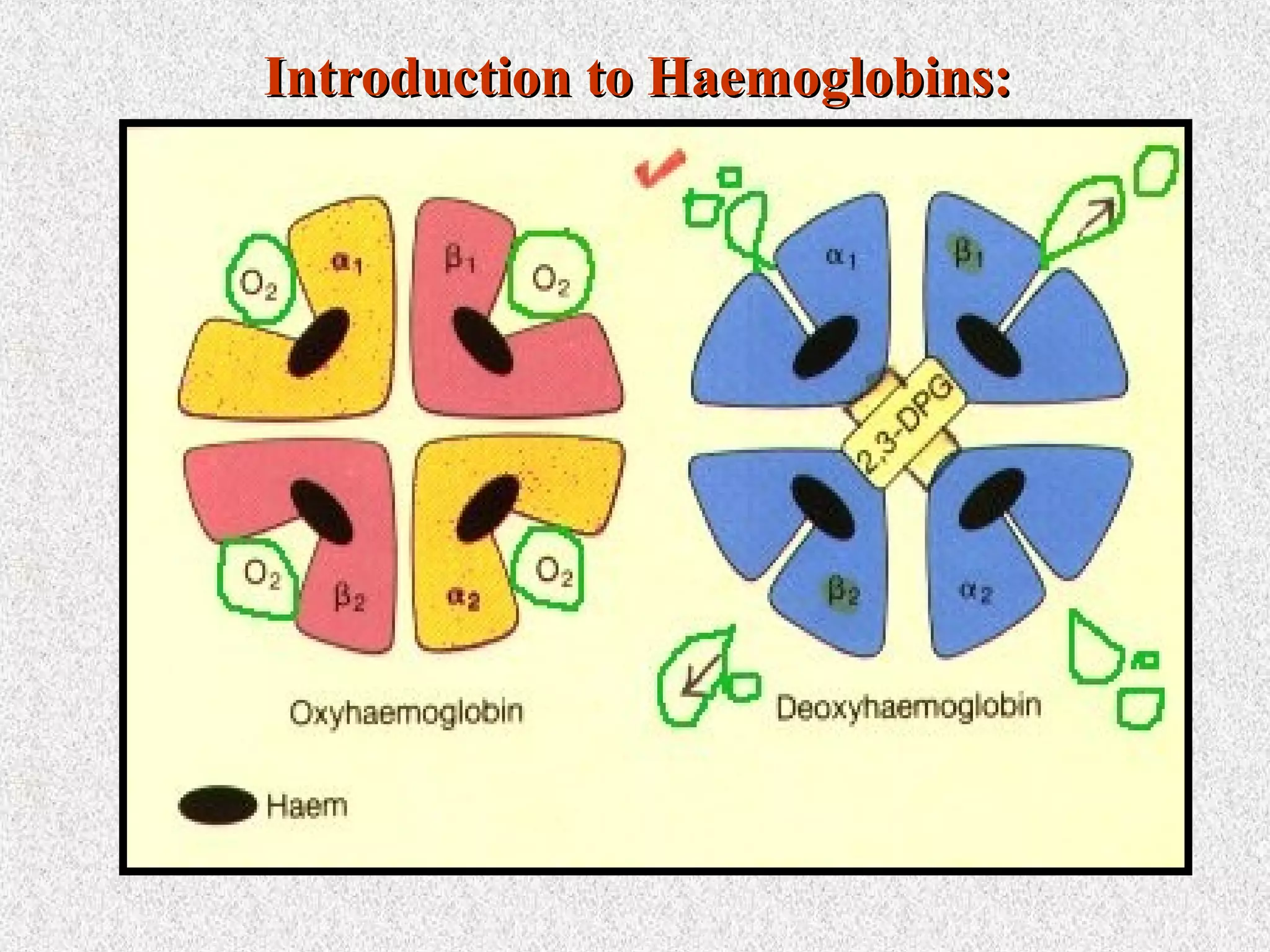







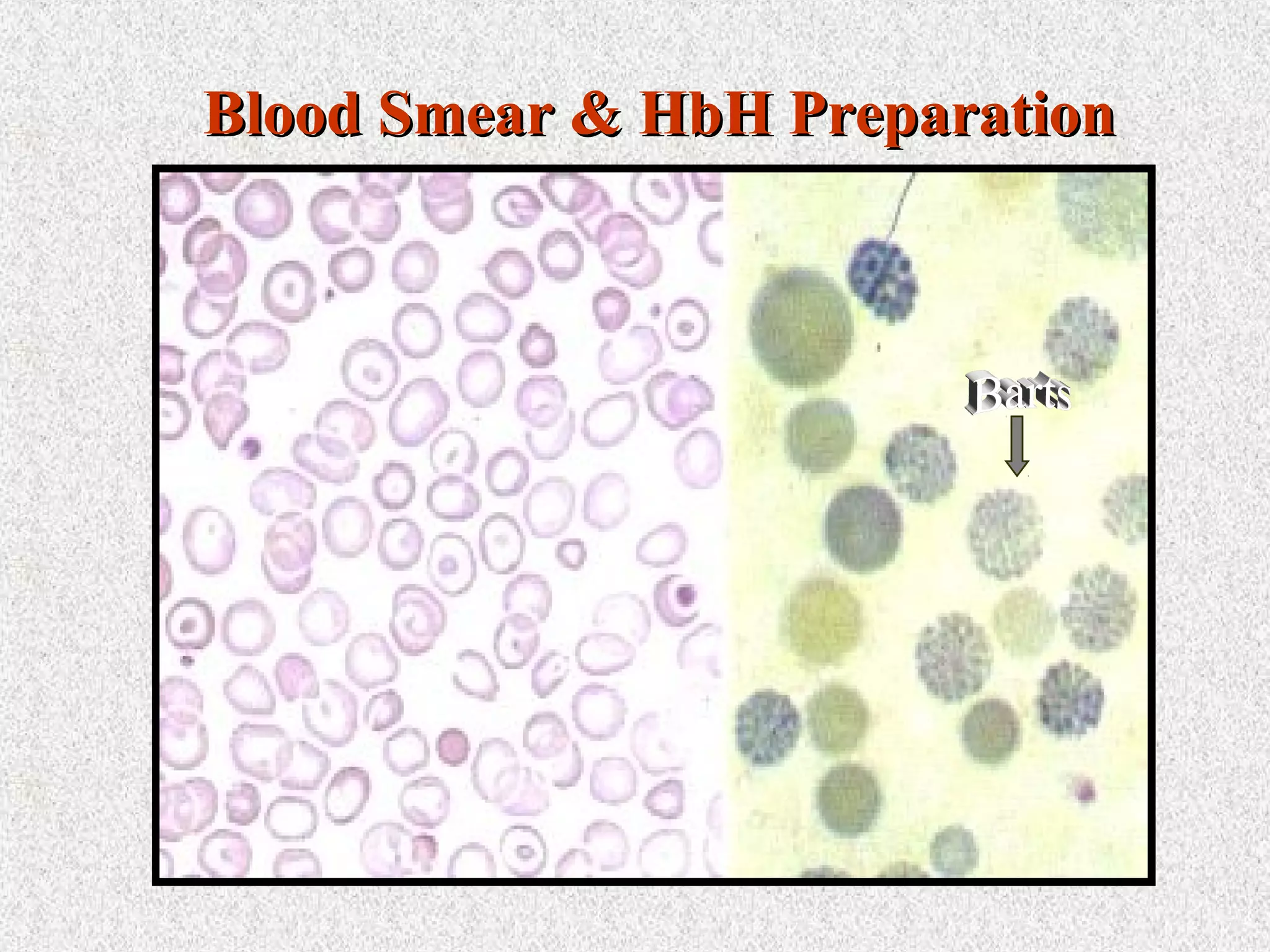




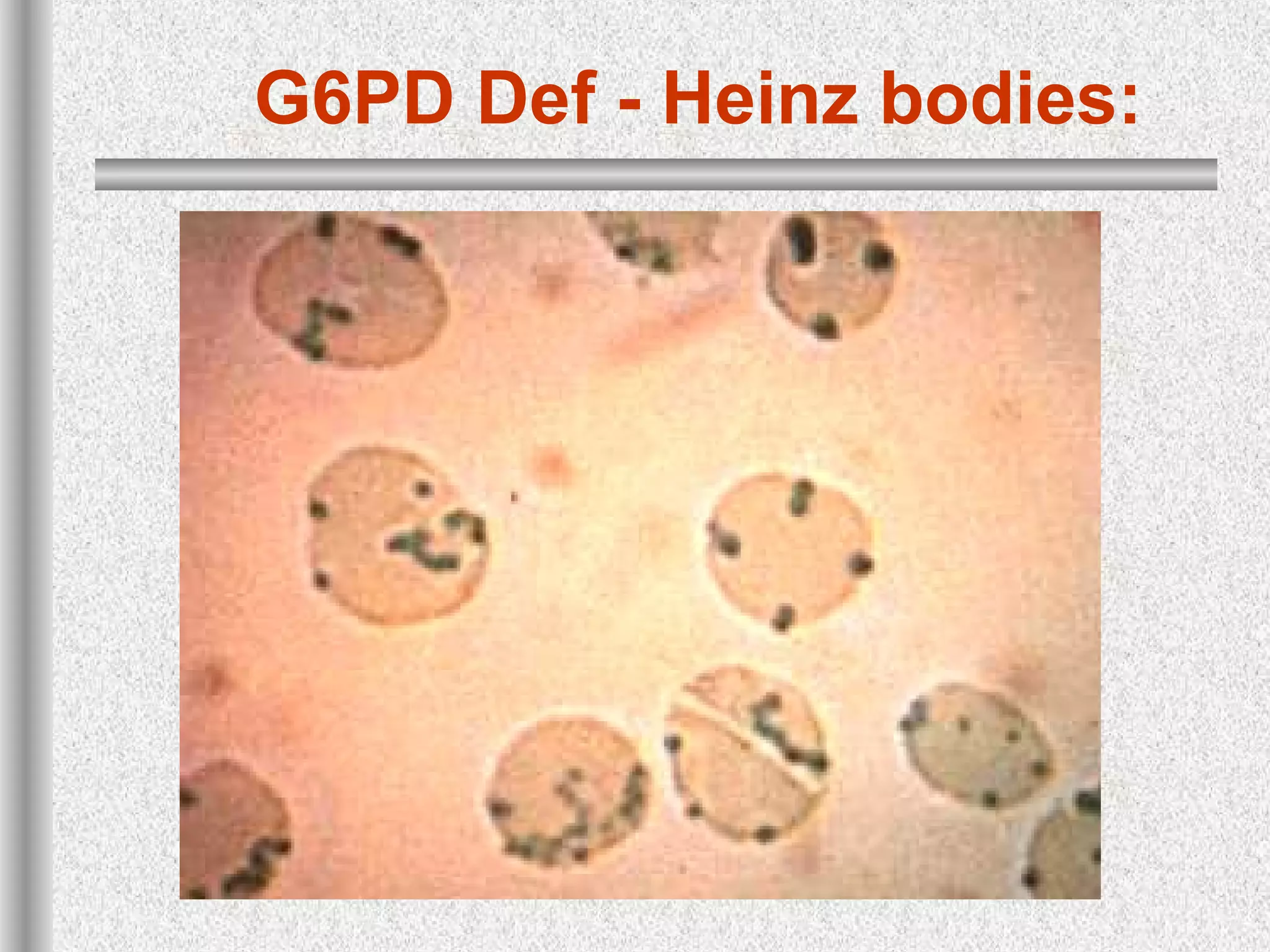


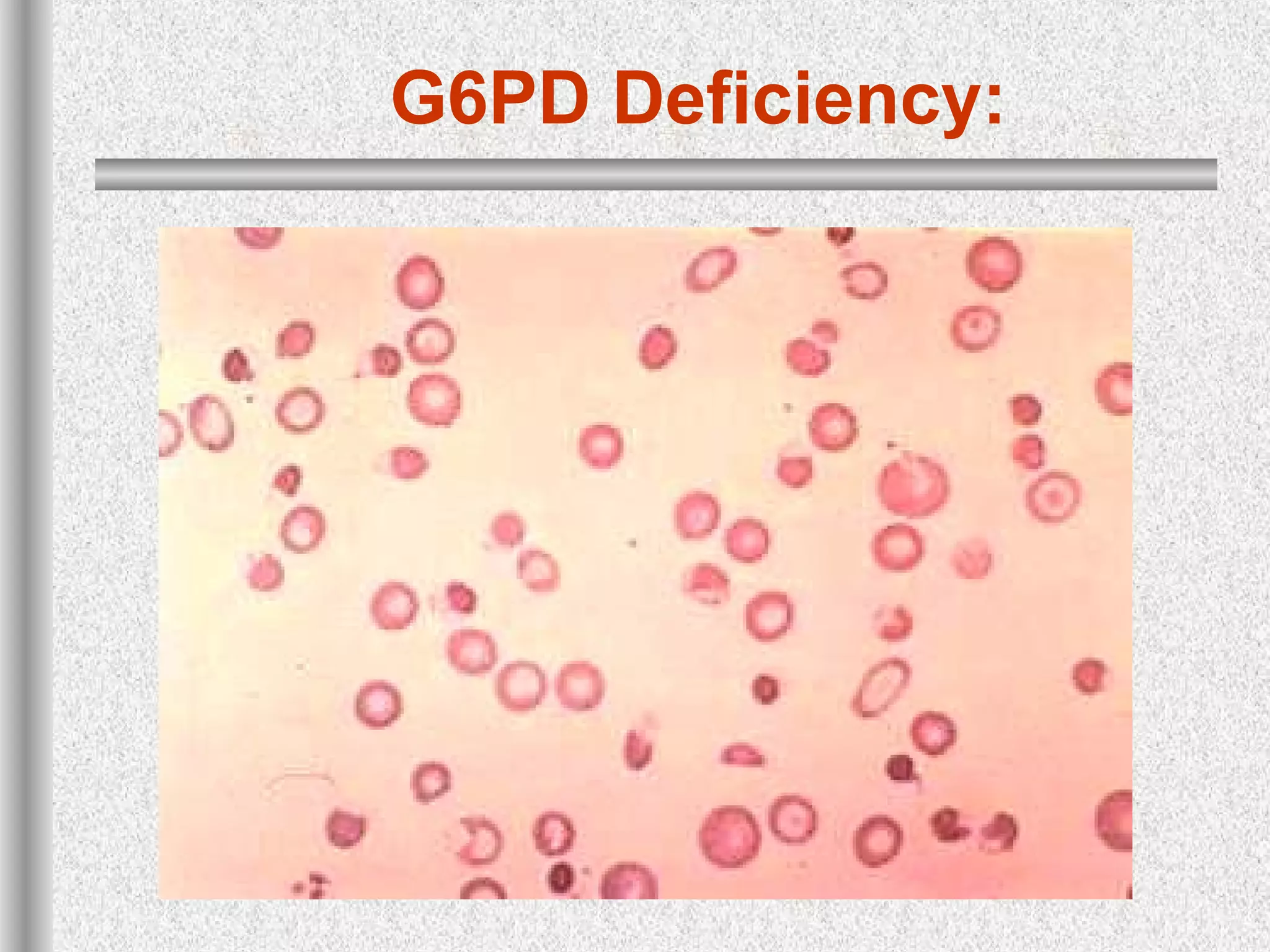


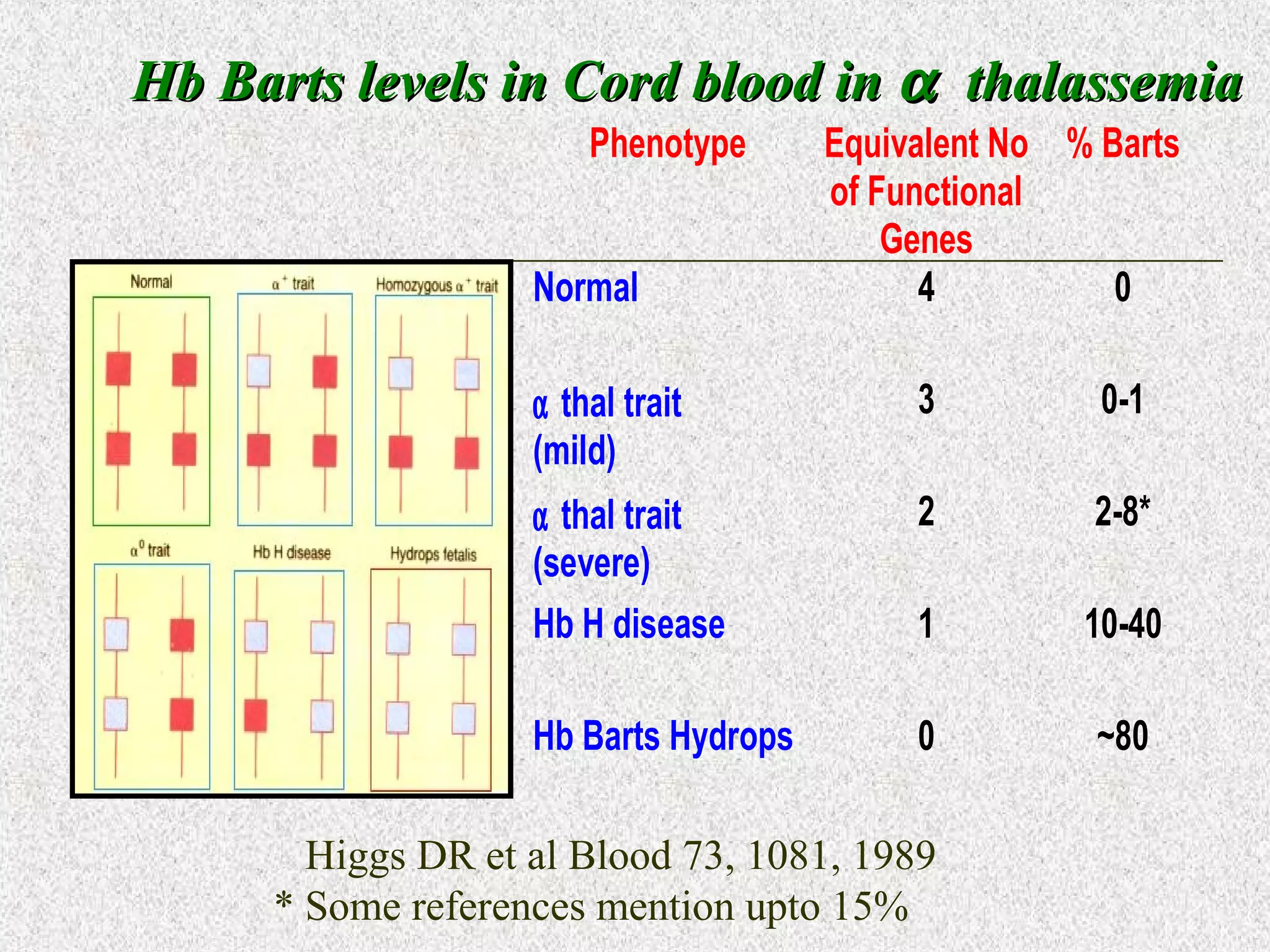

This document discusses hematological disorders including hemolytic anemias and congenital red blood cell disorders. It provides details on laboratory tests used to evaluate anemias, such as blood smears, hemoglobin electrophoresis, and enzyme assays. Specific disorders covered include thalassemias, sickle cell disease, hereditary spherocytosis, elliptocytosis, and G6PD deficiency. Thalassemias result from decreased alpha or beta globin chain production and are classified based on severity from hydrops fetalis to thalassemia minor. Sickle cell disease is caused by a mutation in the beta globin gene.































































































