Downloaded 1,227 times






















![Hyperosmolar hyperglycaemic nonketotic
syndrome
• Precipitating factors include
• Acute infections and other medical conditions
• Drugs that impair glucose tolerance (glucocorticoids) or increase fluid loss (diuretics)
• Non-adherence to diabetes treatment
• Serum ketones are not present
• type 2 DM produce enough insulin to suppress ketogenesis.
• Absence of Ketones - significantly longer period of osmotic dehydration
before presentation, therefore
• plasma glucose (> 33 mmol/L])
• osmolality (> 320 mOsm/L) are
• typically much higher than in diabetic ketoacidosis (DKA)](https://image.slidesharecdn.com/endocrineemergenciesworkingcopy97-140604011033-phpapp02/85/Endocrine-Emergencies-24-320.jpg)












![Congenital adrenal insufficiency
• The most common forms, 21-hydroxylase deficiency and 11β-
hydroxylase deficiency, precursors proximal to the enzyme block
accumulate and are shunted into adrenal androgens.
• The consequent excess androgen secretion causes varying degrees of
virilization in external genitals of affected female fetuses; no defects
are discernible in external genitals of male fetuses.
• In some less common forms affecting enzymes other than 21-
hydroxylase and 11β-hydroxylase, the enzyme block impairs
androgen synthesis (dehydoepiandrosterone [DHEAS] or
androstenedione).
• As a result, virilization of male fetuses is inadequate, but no defect is
discernible in female fetuses.](https://image.slidesharecdn.com/endocrineemergenciesworkingcopy97-140604011033-phpapp02/85/Endocrine-Emergencies-38-320.jpg)




























![Central Diabetes insipidus
• Deficiency of vasopressin (ADH) due to a
hypothalamic-pituitary disorder (central
DI [CDI])
• Primary
• Genetic
• Secondary
• hypophysectomy, cranial injuries
(particularly basal skull fractures),
suprasellar and intrasellar tumors (primary
or metastatic),
• Langerhans cell histiocytosis (Hand-
Schüller-Christian disease),
• lymphocytic hypophysitis,
• Granulomas (sarcoidosis or TB)
• Vascular lesions (aneurysm, thrombosis)
• Infections (encephalitis, meningitis](https://image.slidesharecdn.com/endocrineemergenciesworkingcopy97-140604011033-phpapp02/85/Endocrine-Emergencies-73-320.jpg)


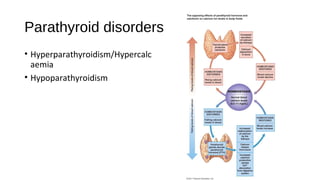








This document discusses various endocrine emergencies, including: - Glucose metabolism disorders like hypoglycemia, diabetic ketoacidosis, and hyperglycemic hyperosmolar nonketotic syndrome. - Adrenal disorders such as Addison's disease, adrenal crisis, and congenital adrenal insufficiency. - It provides details on the causes, pathophysiology, symptoms, diagnosis, and treatment of these various conditions. The focus is on clinically relevant information to recognize and manage endocrine emergencies.









































































































