Downloaded 1,570 times



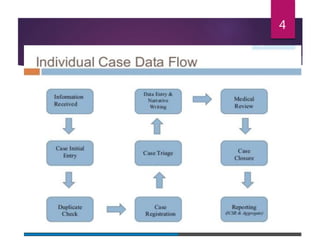
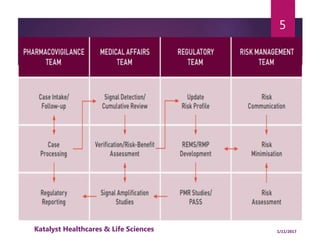







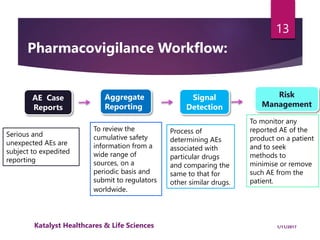
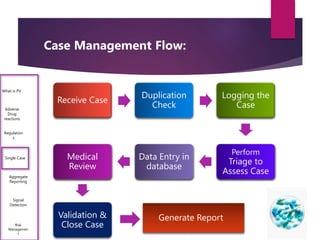
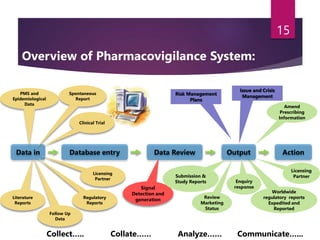




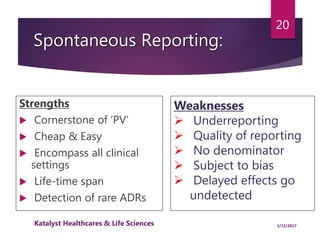

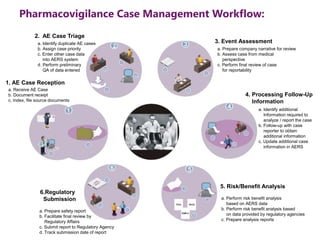



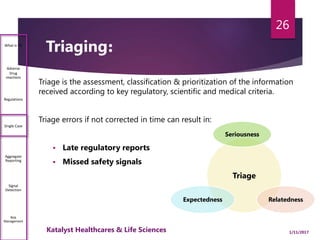

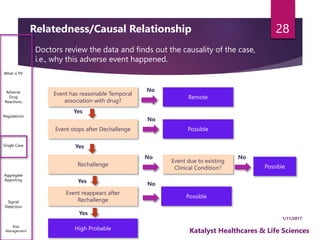



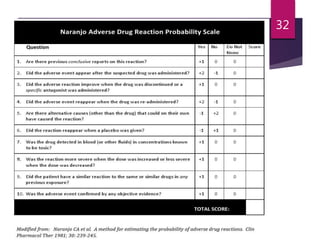






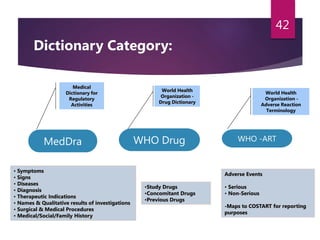



































The document outlines the pharmacovigilance process, focusing on key elements such as case validity, reporting requirements, and the workflow for managing adverse drug reactions. It discusses the minimum data elements required for reporting, methods for assessing causality, and the importance of aggregate reporting and signal detection. Additionally, it covers the types of safety reports and regulatory timelines for reporting adverse events.











































































































