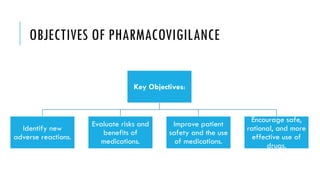
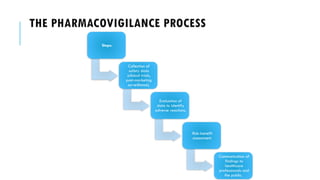
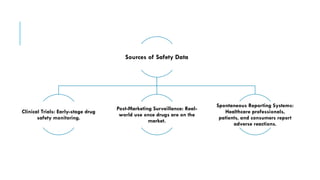
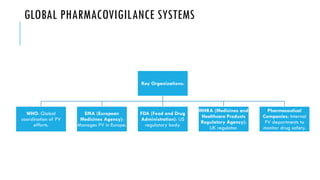
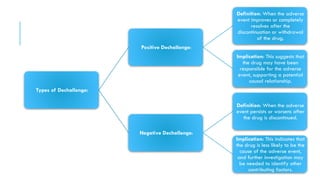
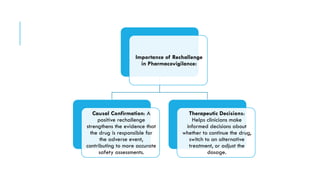
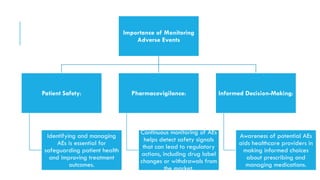
The document outlines the importance and objectives of pharmacovigilance, which involves monitoring drug safety and adverse effects to protect patient health and improve medication efficacy. It details methods for identifying adverse reactions, the regulatory frameworks governing pharmacovigilance, and the roles of various agencies in ensuring drug safety through data collection, analysis, and reporting. The document emphasizes the significance of risk assessment, management strategies, and the processes for validating adverse event reports to inform clinical practice and regulatory actions.