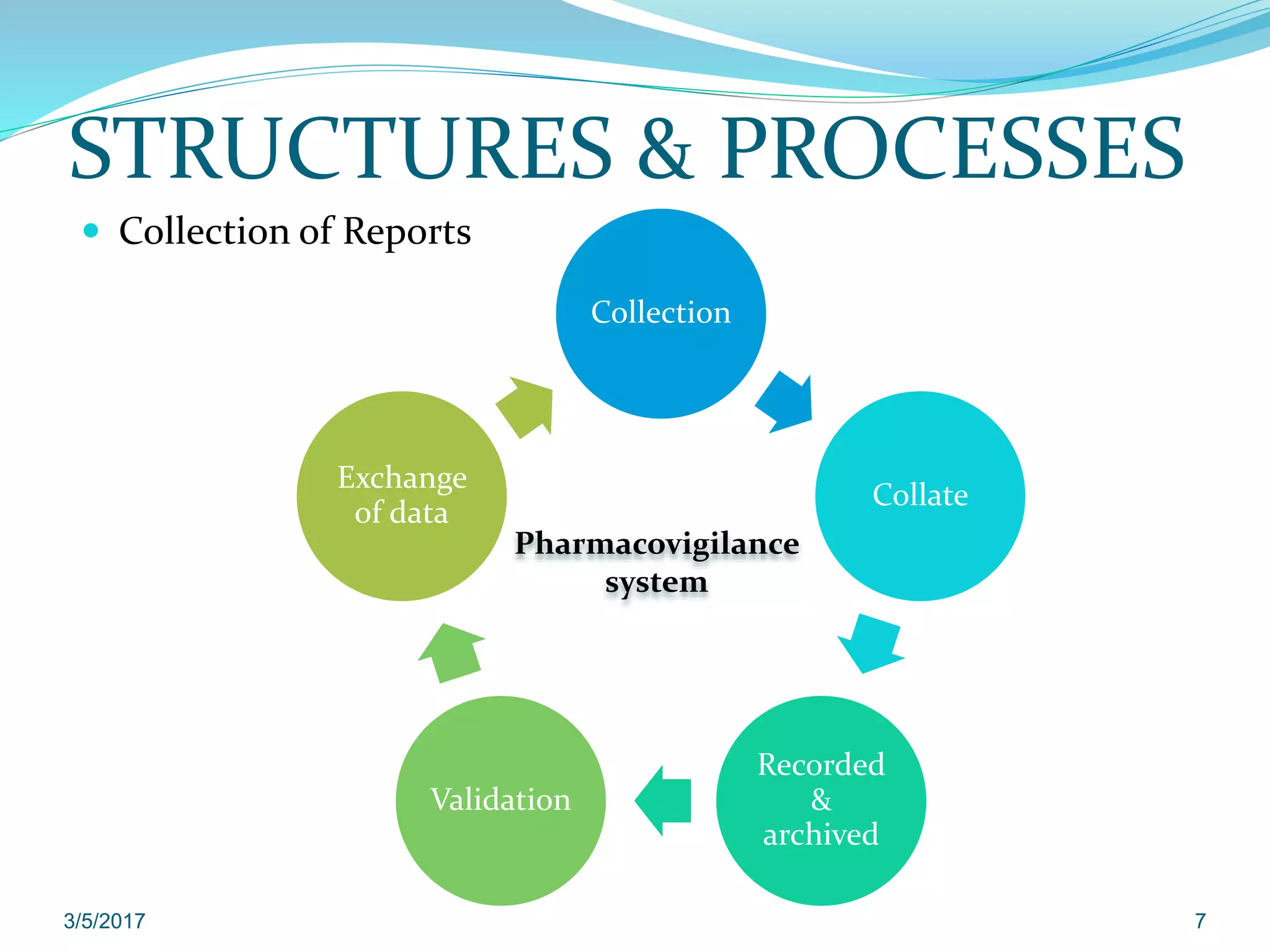
The document provides guidelines on good pharmacovigilance practices for managing and reporting adverse reactions to medicinal products, with an effective date of September 16, 2014. It covers various aspects including terminology related to medicinal products, adverse reactions, and serious adverse reactions, as well as structures and processes for collecting, validating, and managing reports. Special situations are addressed, including those involving pregnancy, pediatric and elderly populations, along with specific reporting time frames and modalities for individual case safety reports.