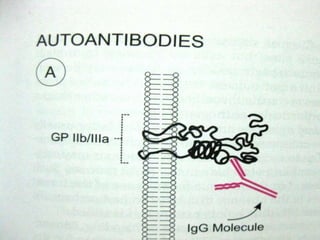
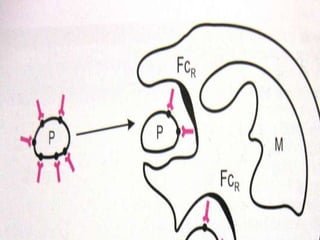
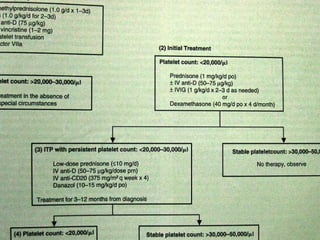
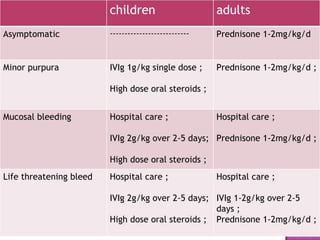
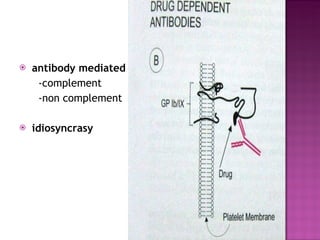
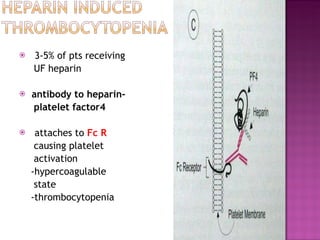
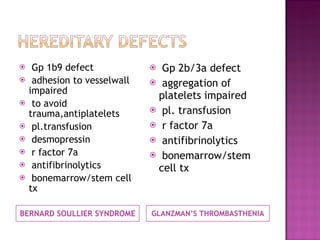
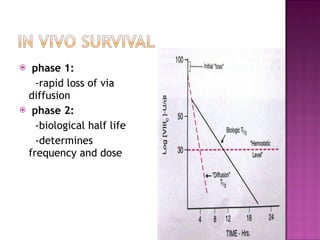
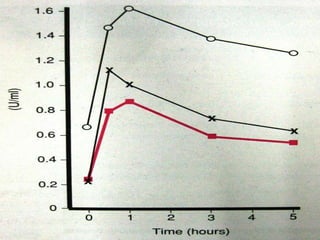
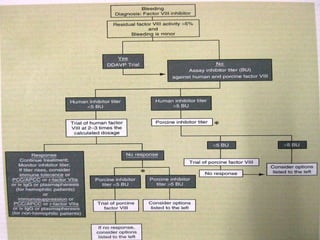
This document summarizes various causes and management of thrombocytopenia and bleeding disorders. It discusses evaluation and treatment of conditions that cause decreased platelet count or function such as immune thrombocytopenia, thrombotic thrombocytopenic purpura, hemophilia, von Willebrand disease, and liver disease. Treatment options include steroids, IV immunoglobulins, platelet transfusions, desmopressin, coagulation factor concentrates, cryoprecipitate, splenectomy and plasma exchange.