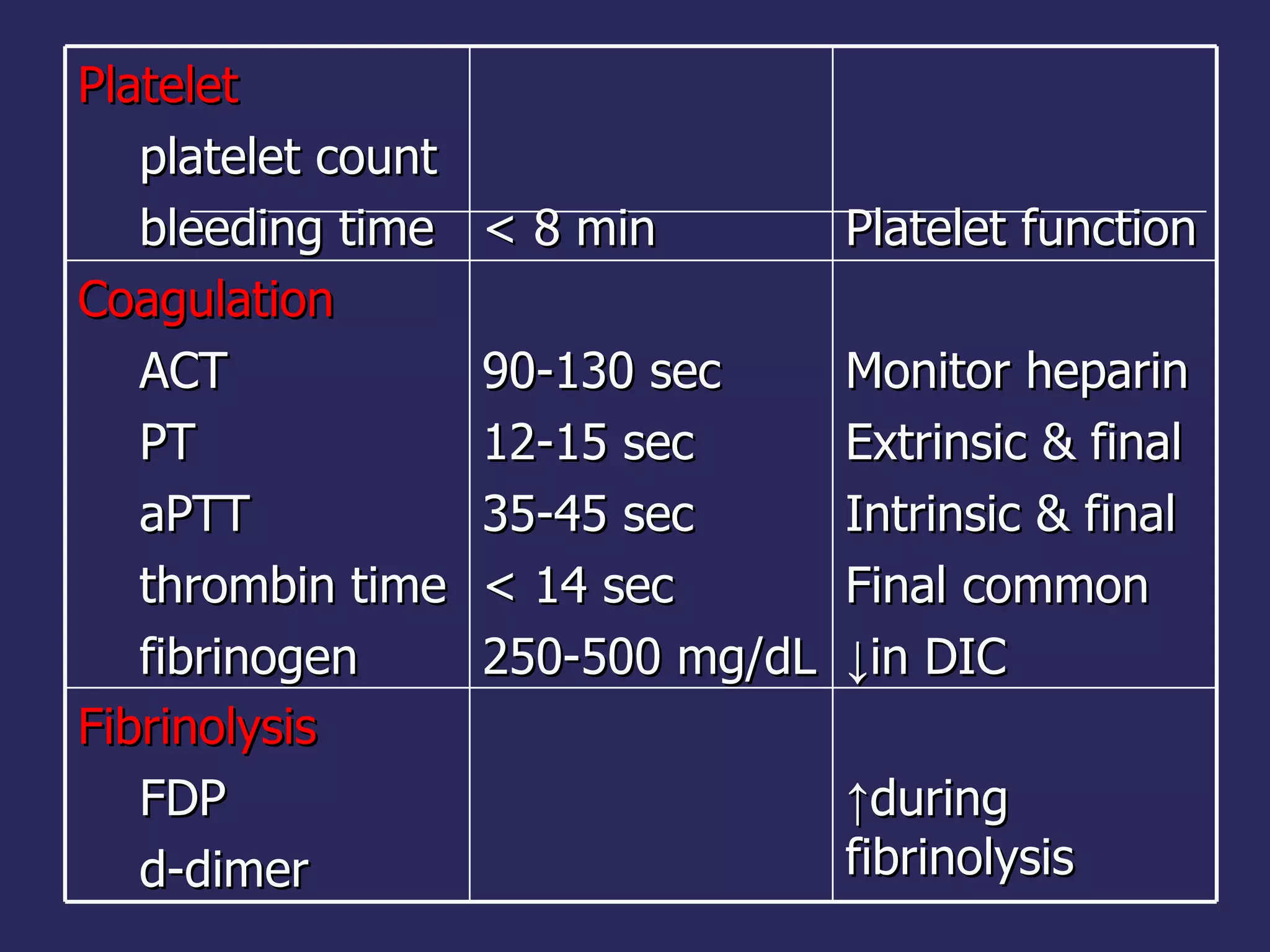
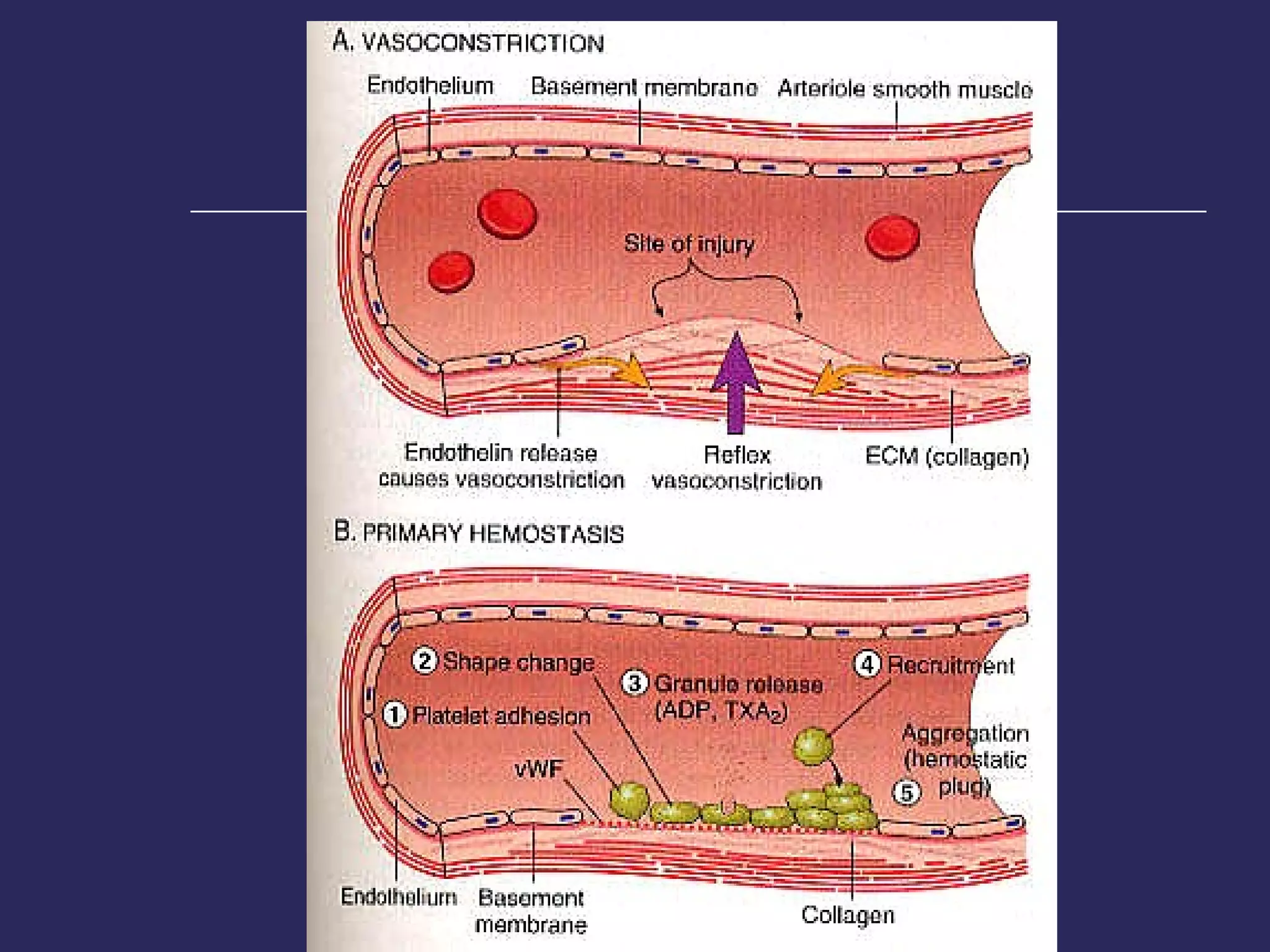
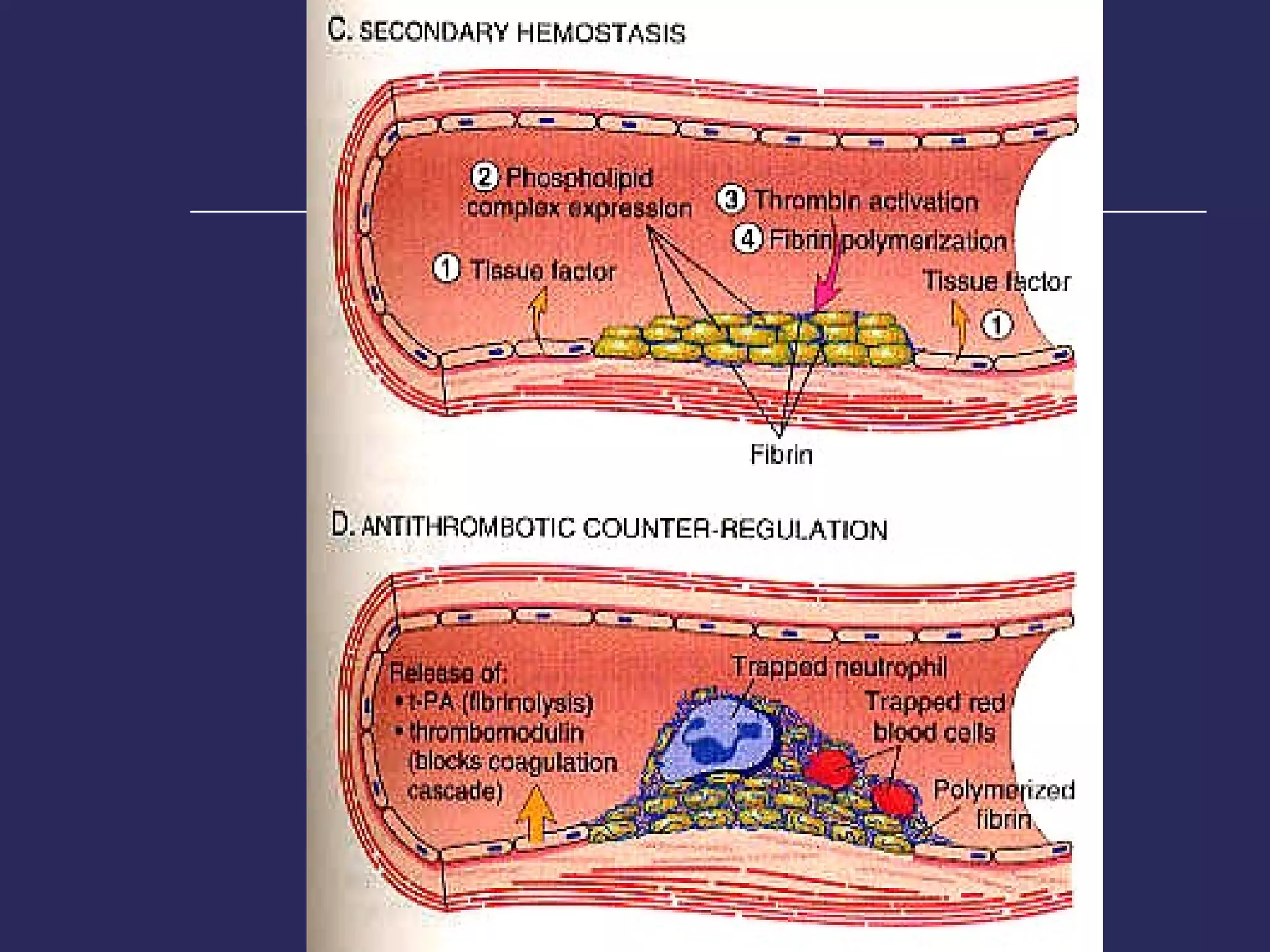
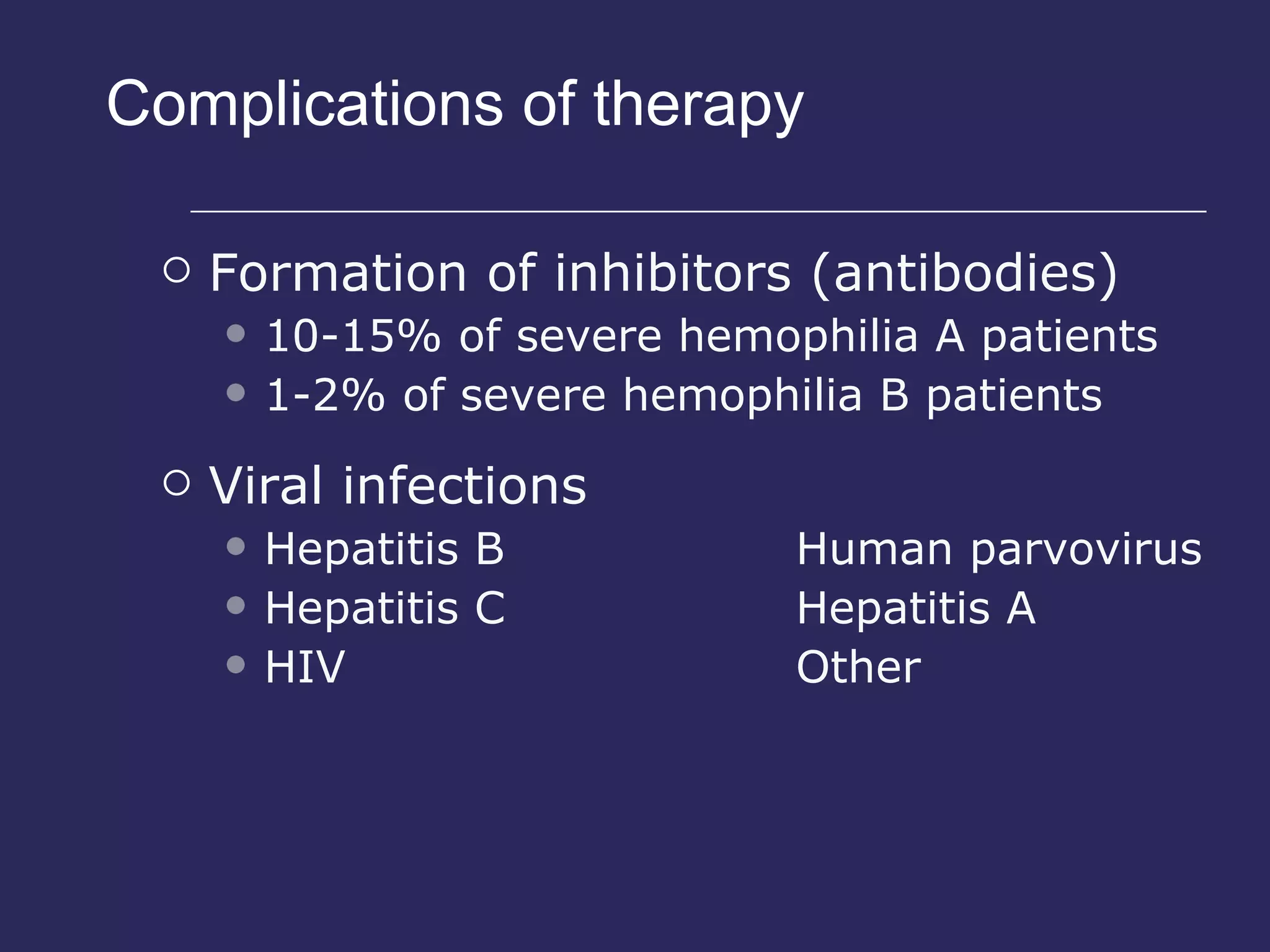
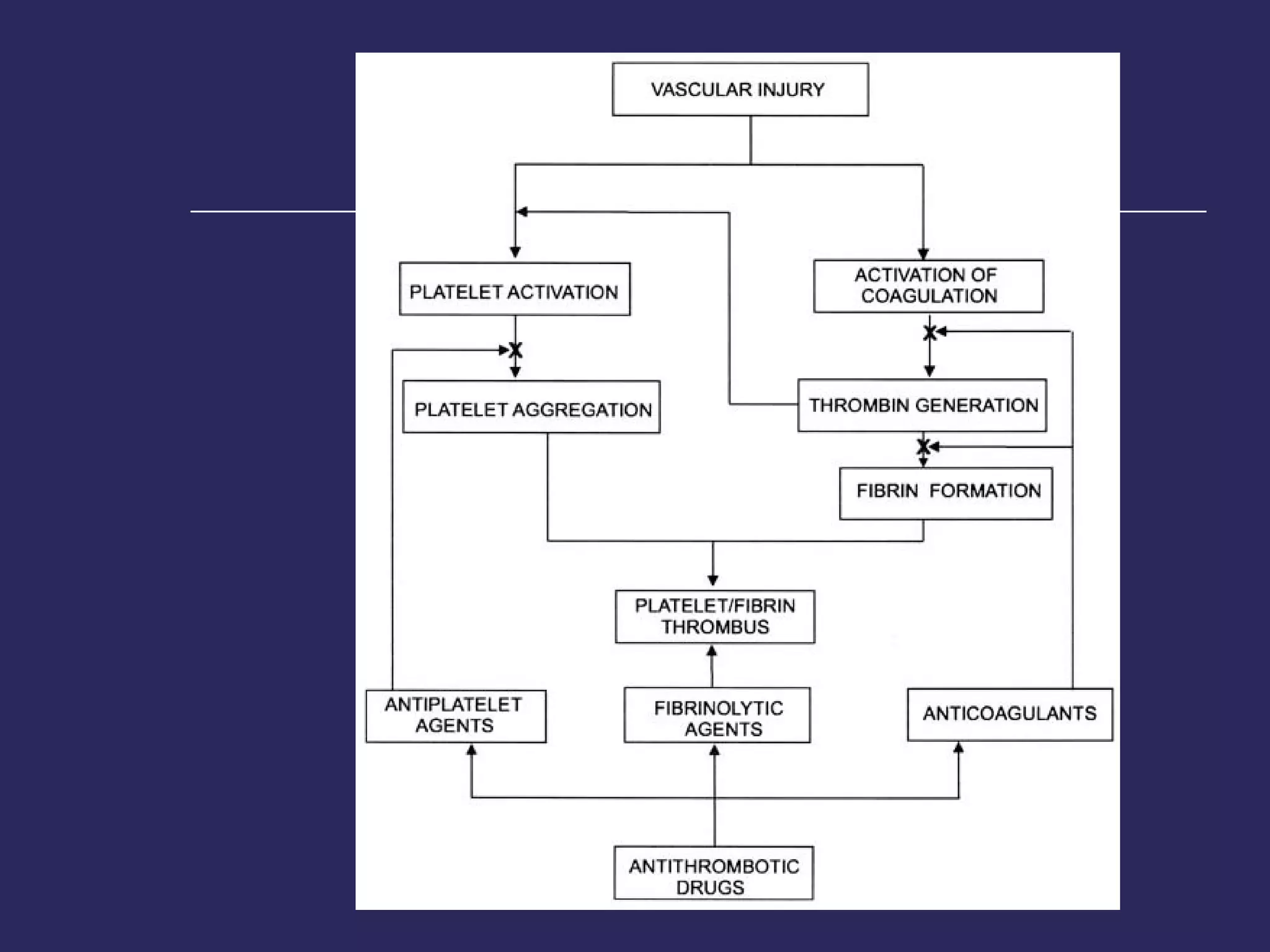
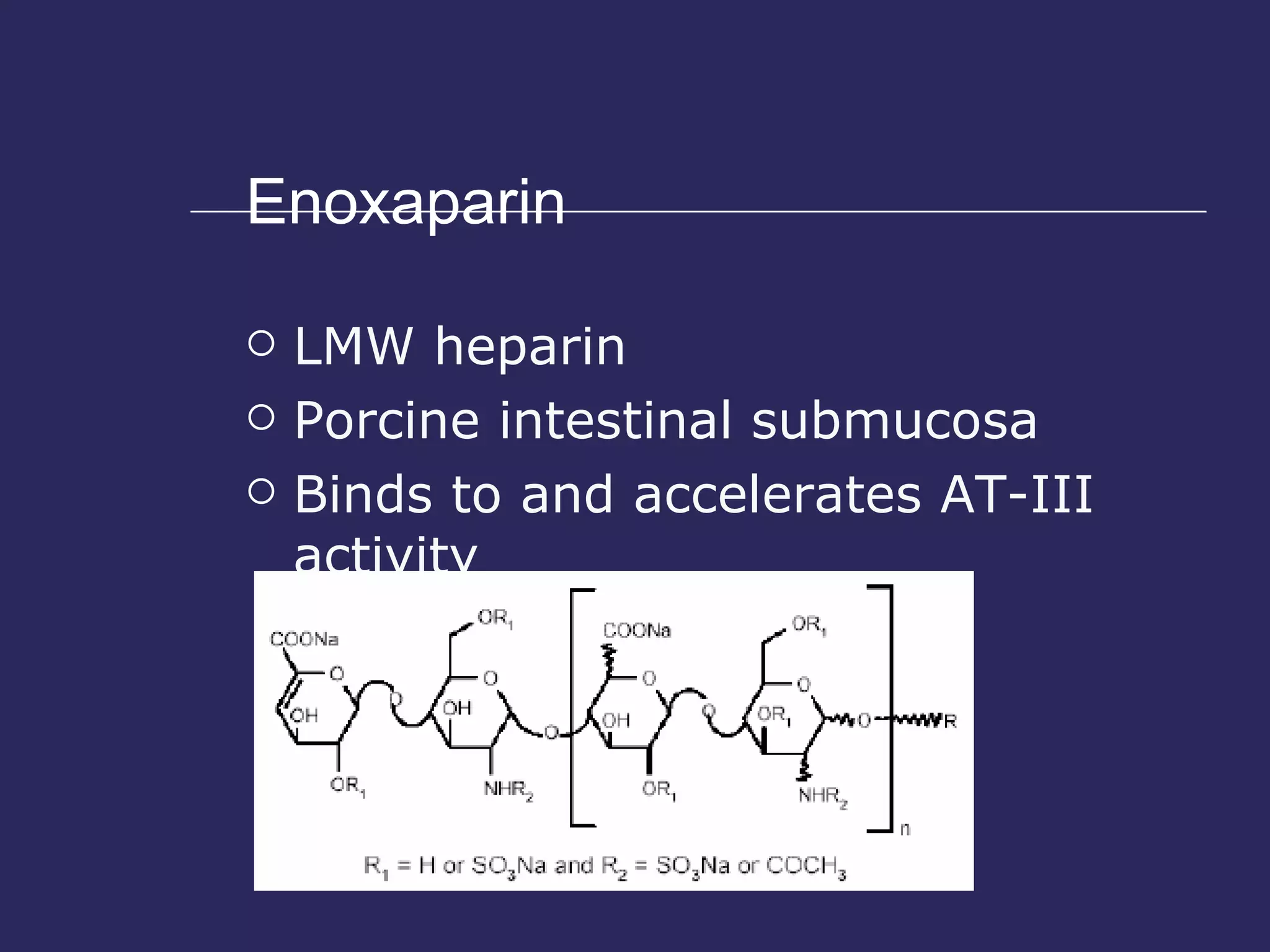
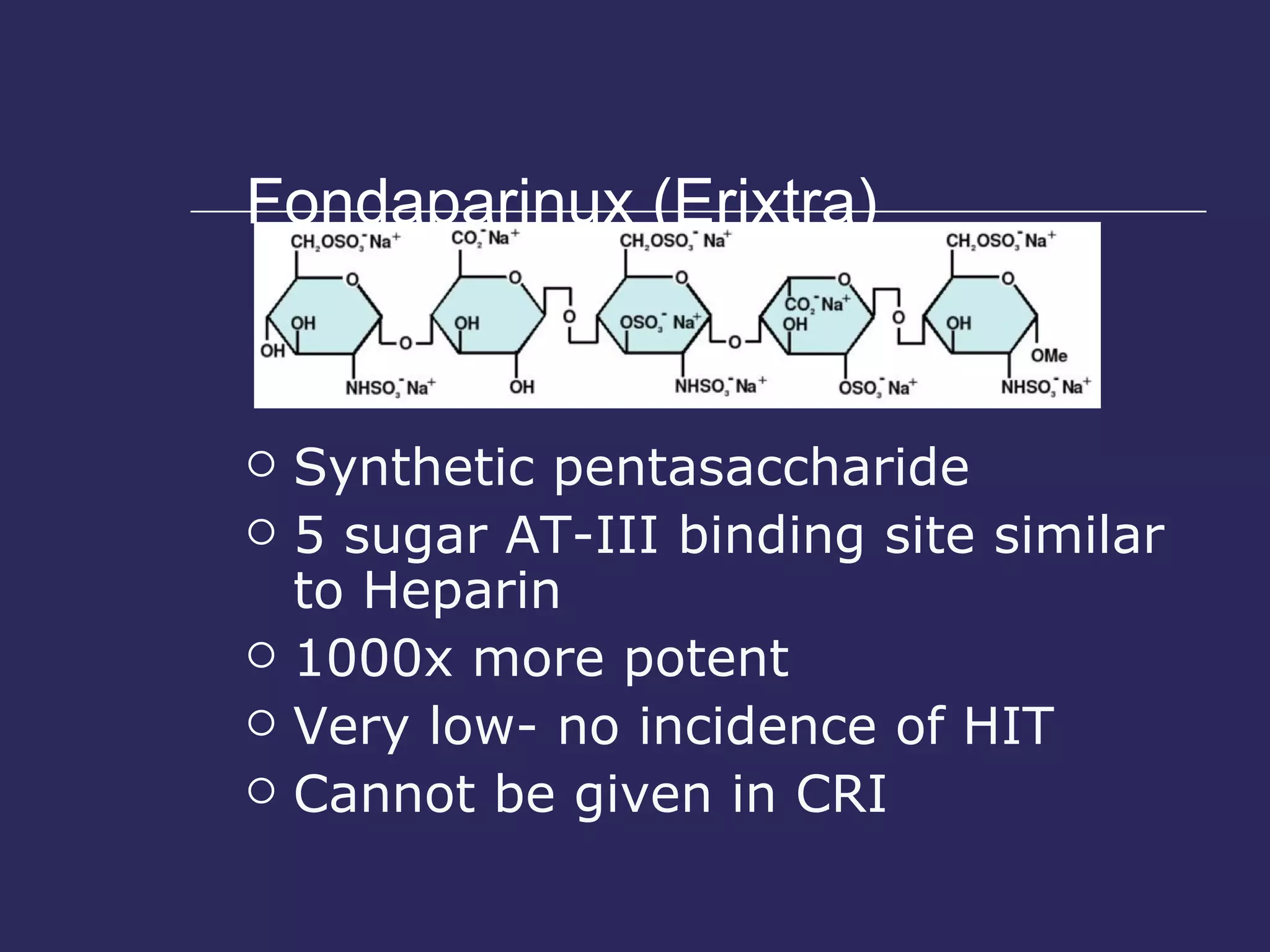
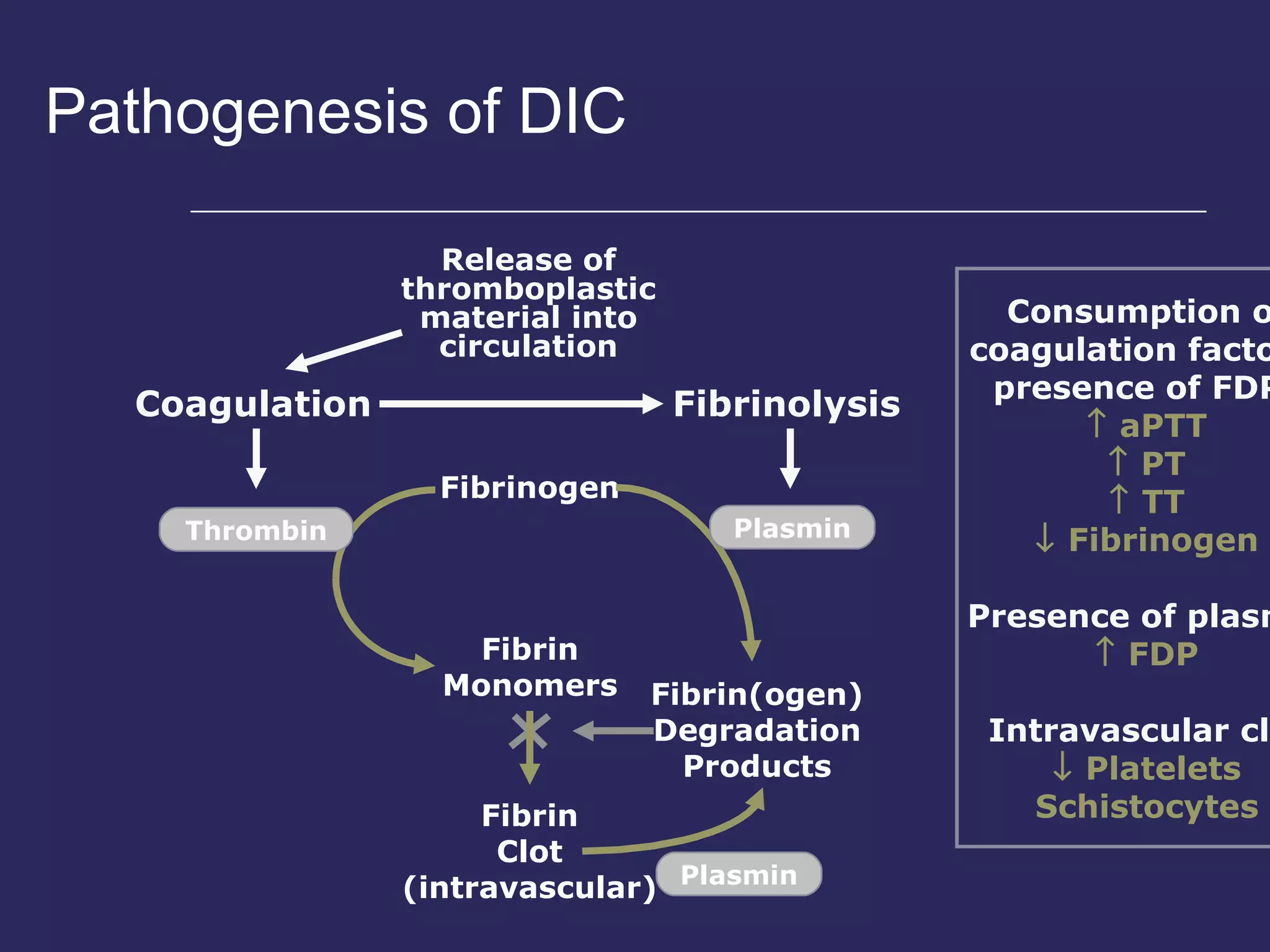
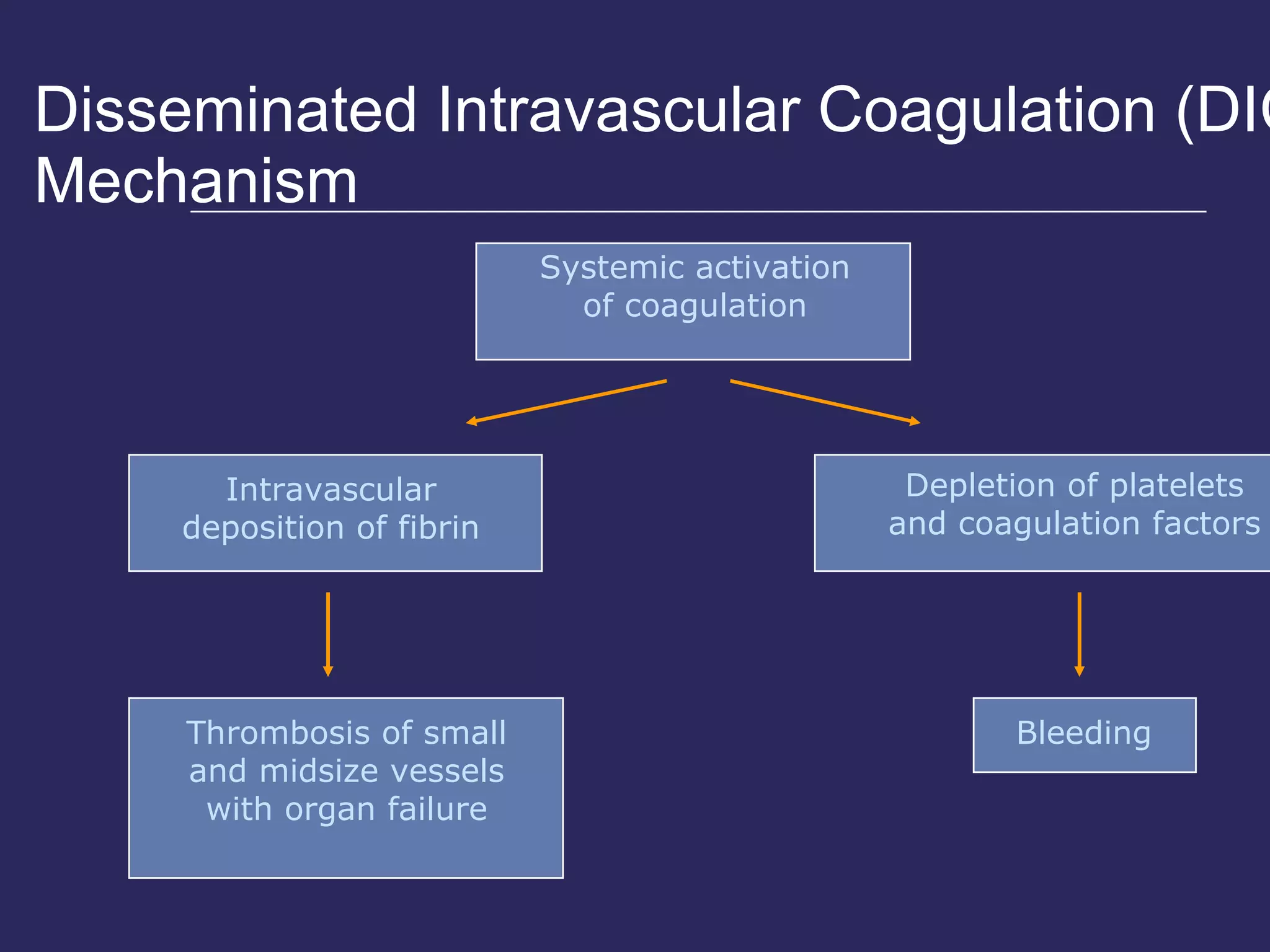
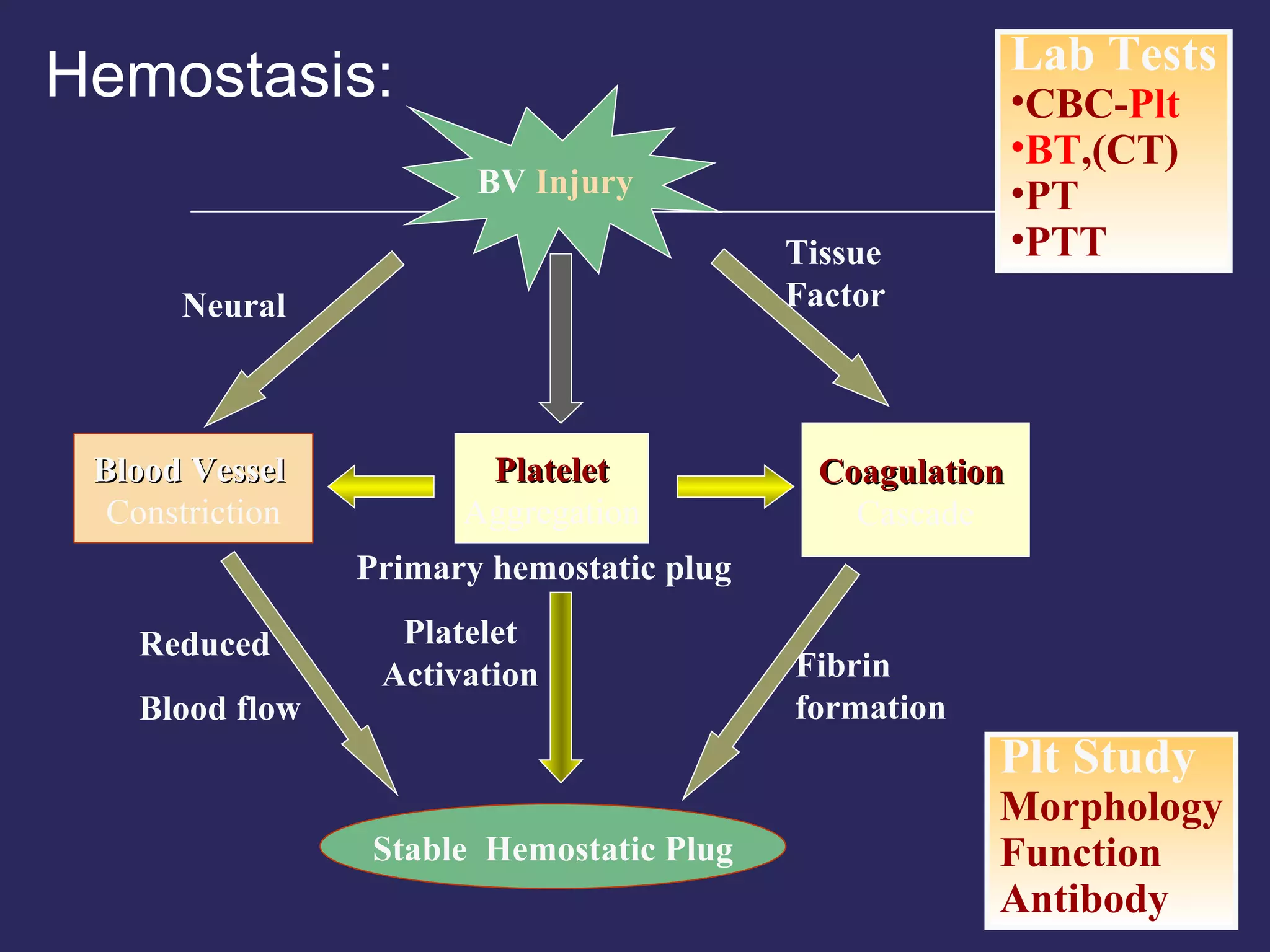
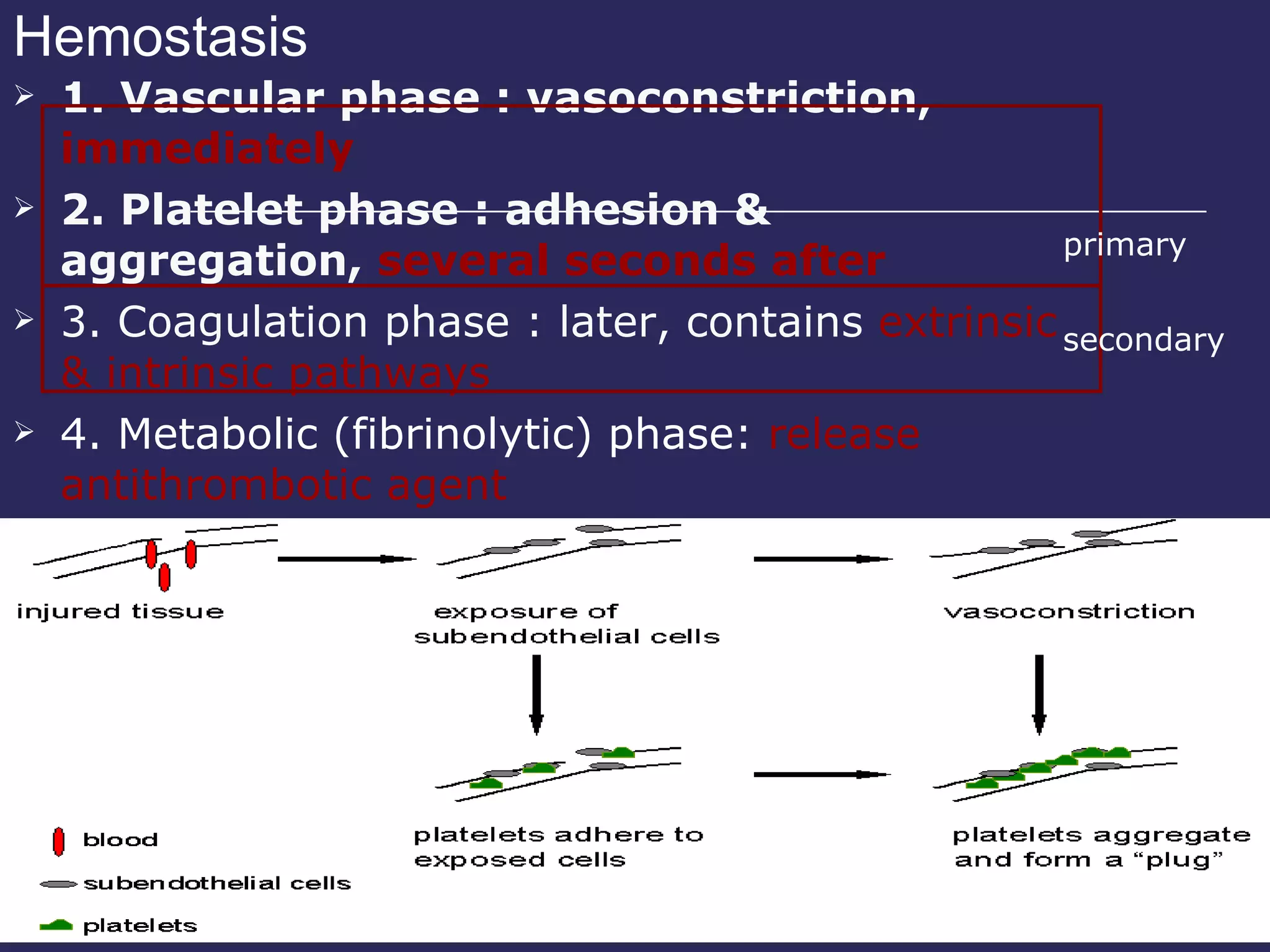
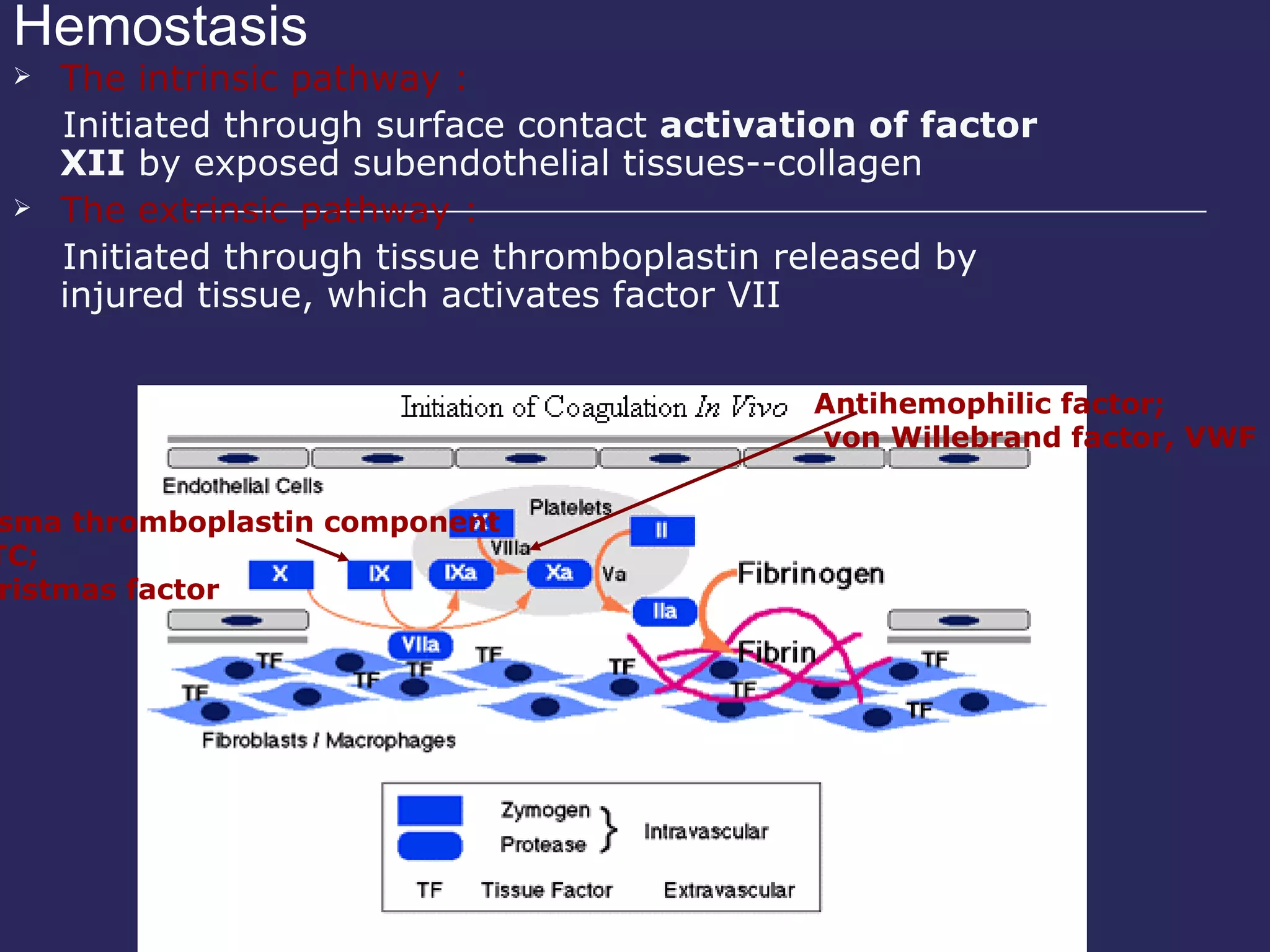
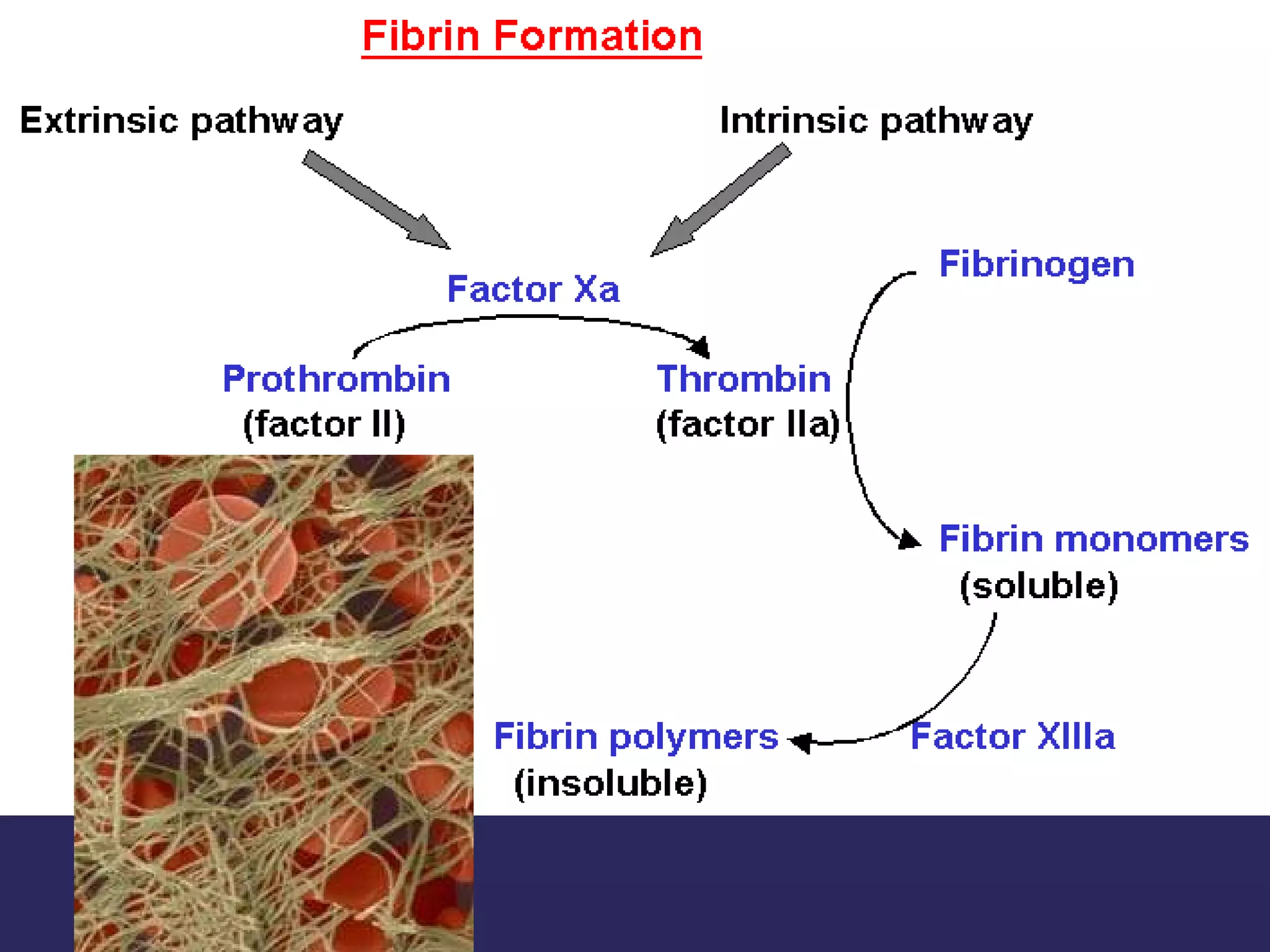
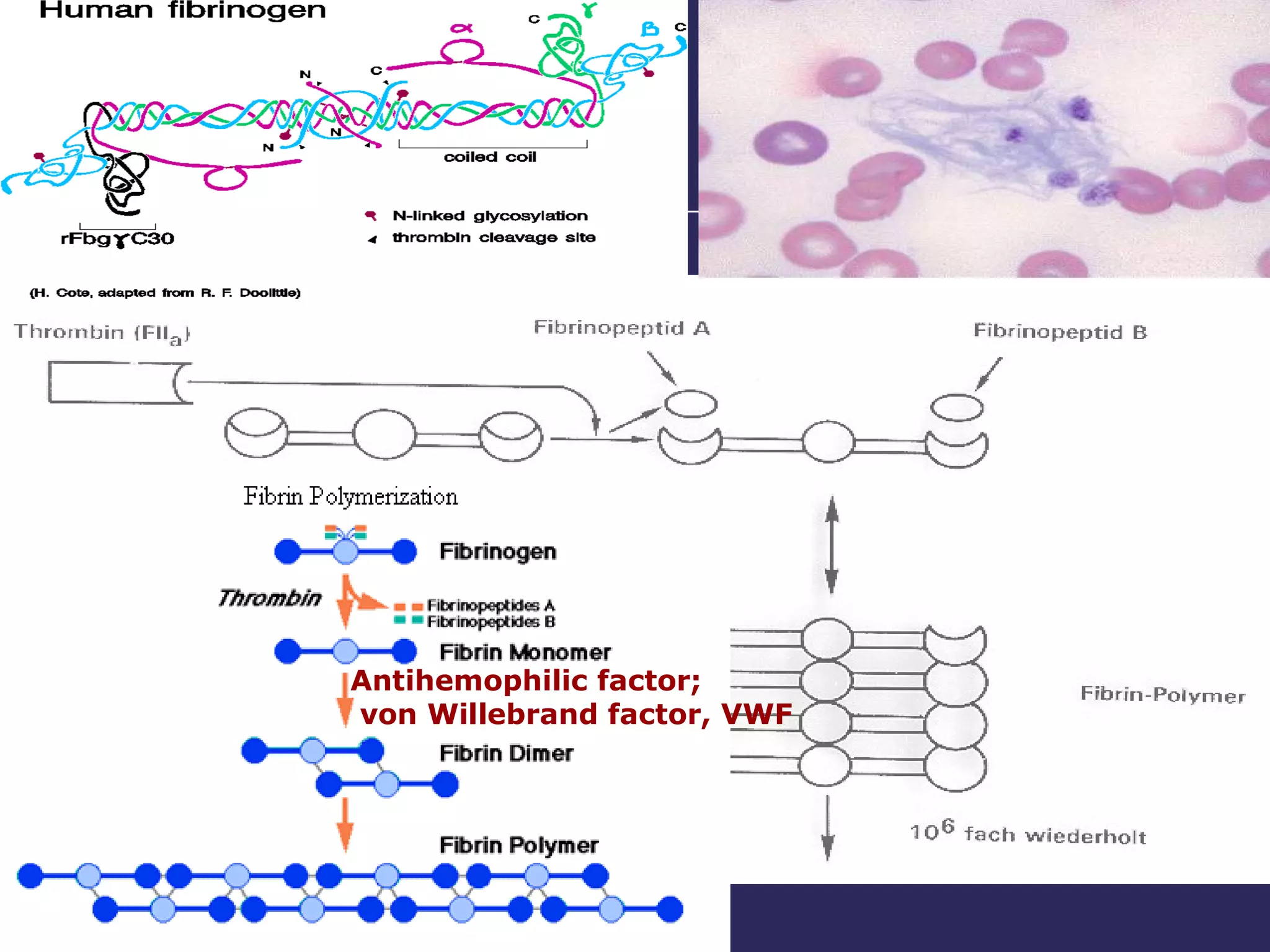
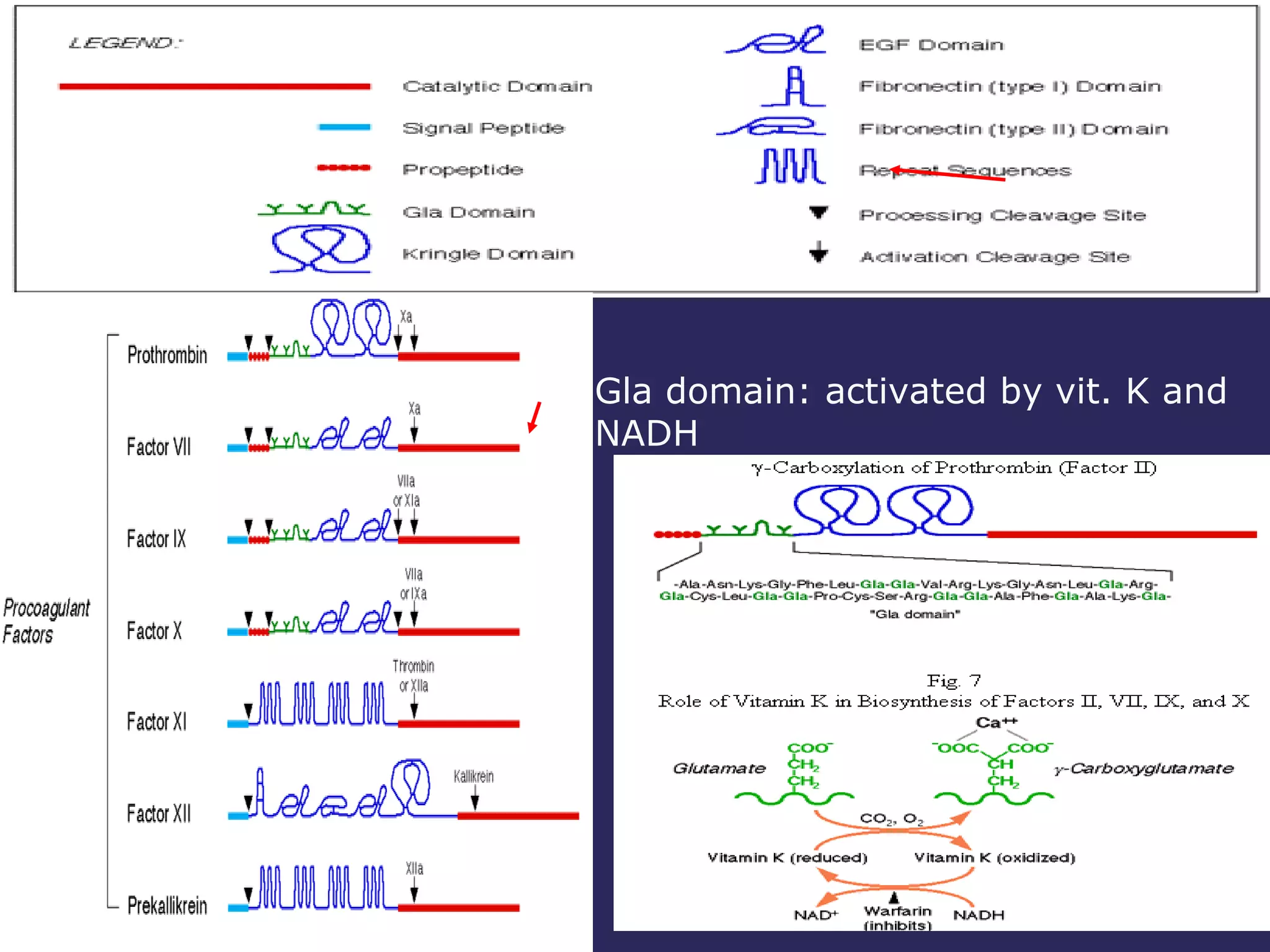
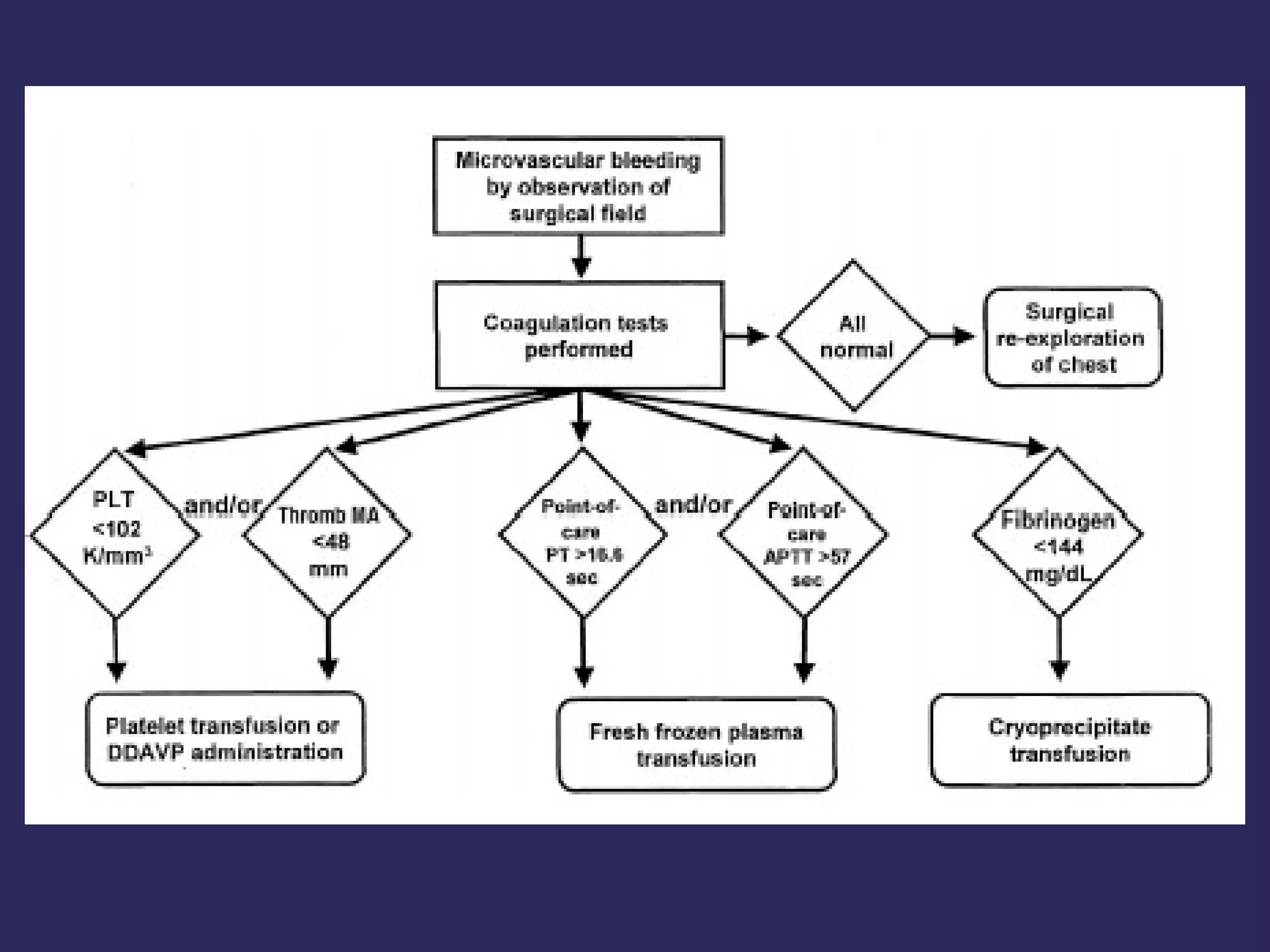
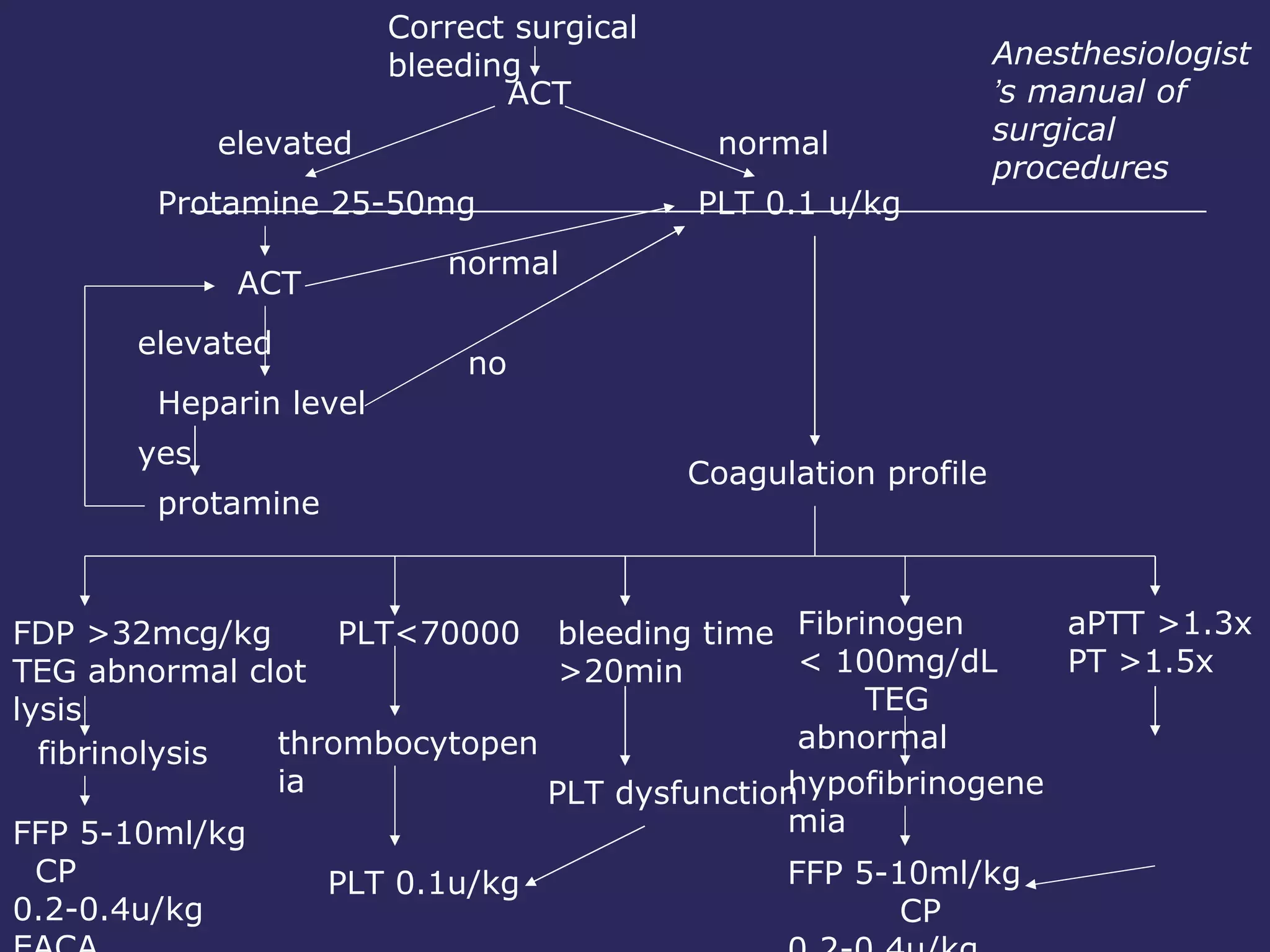
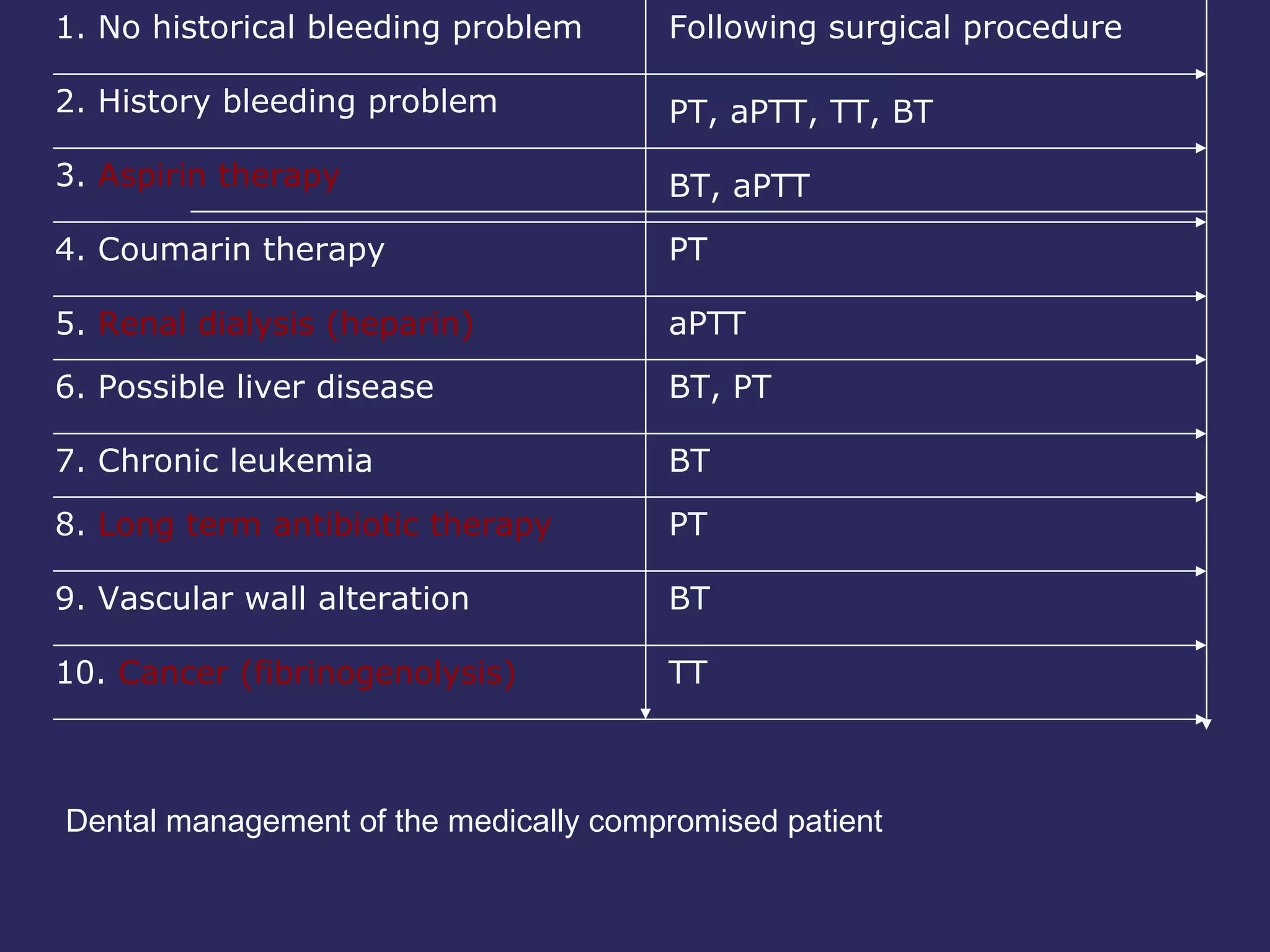
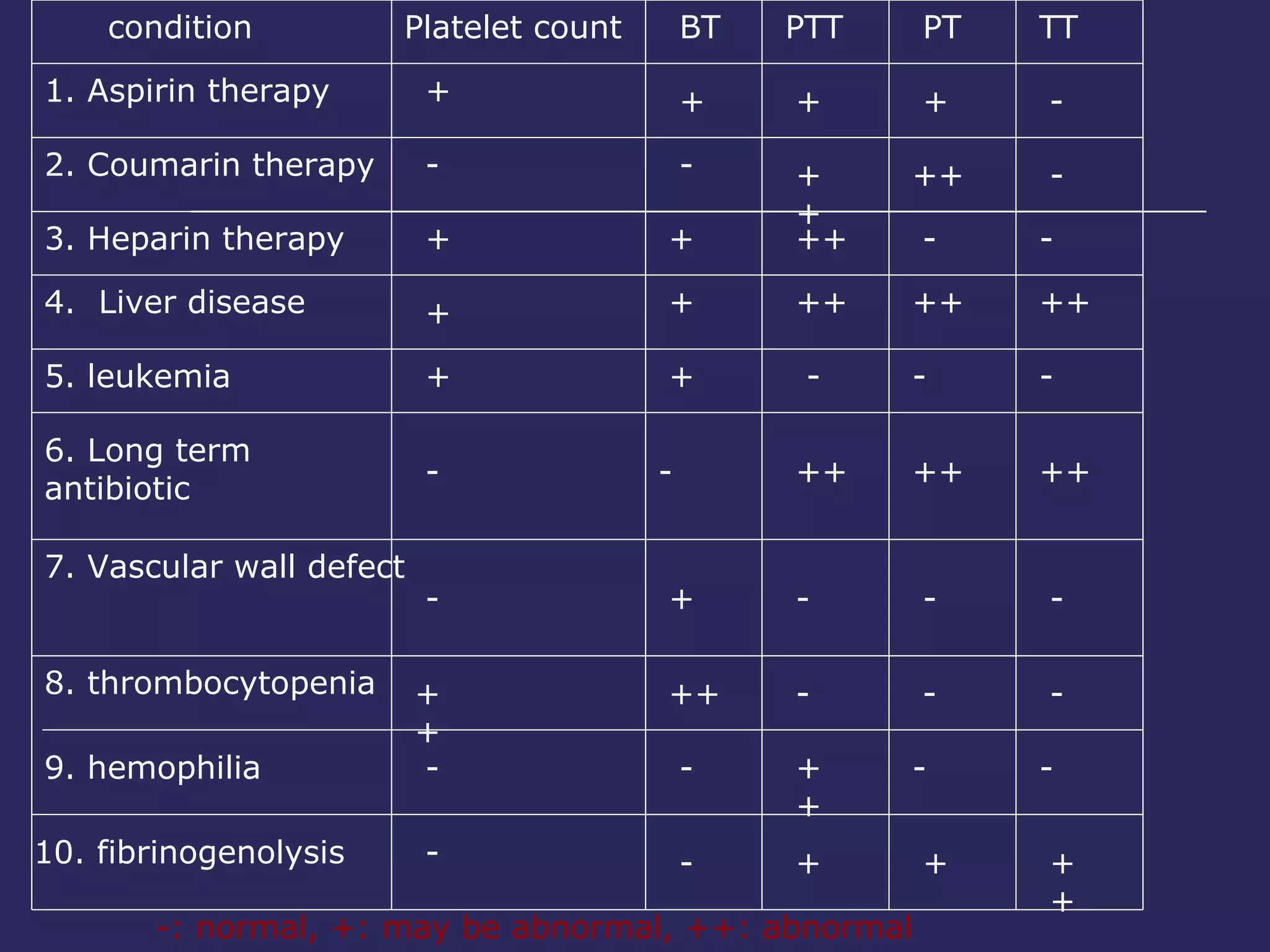
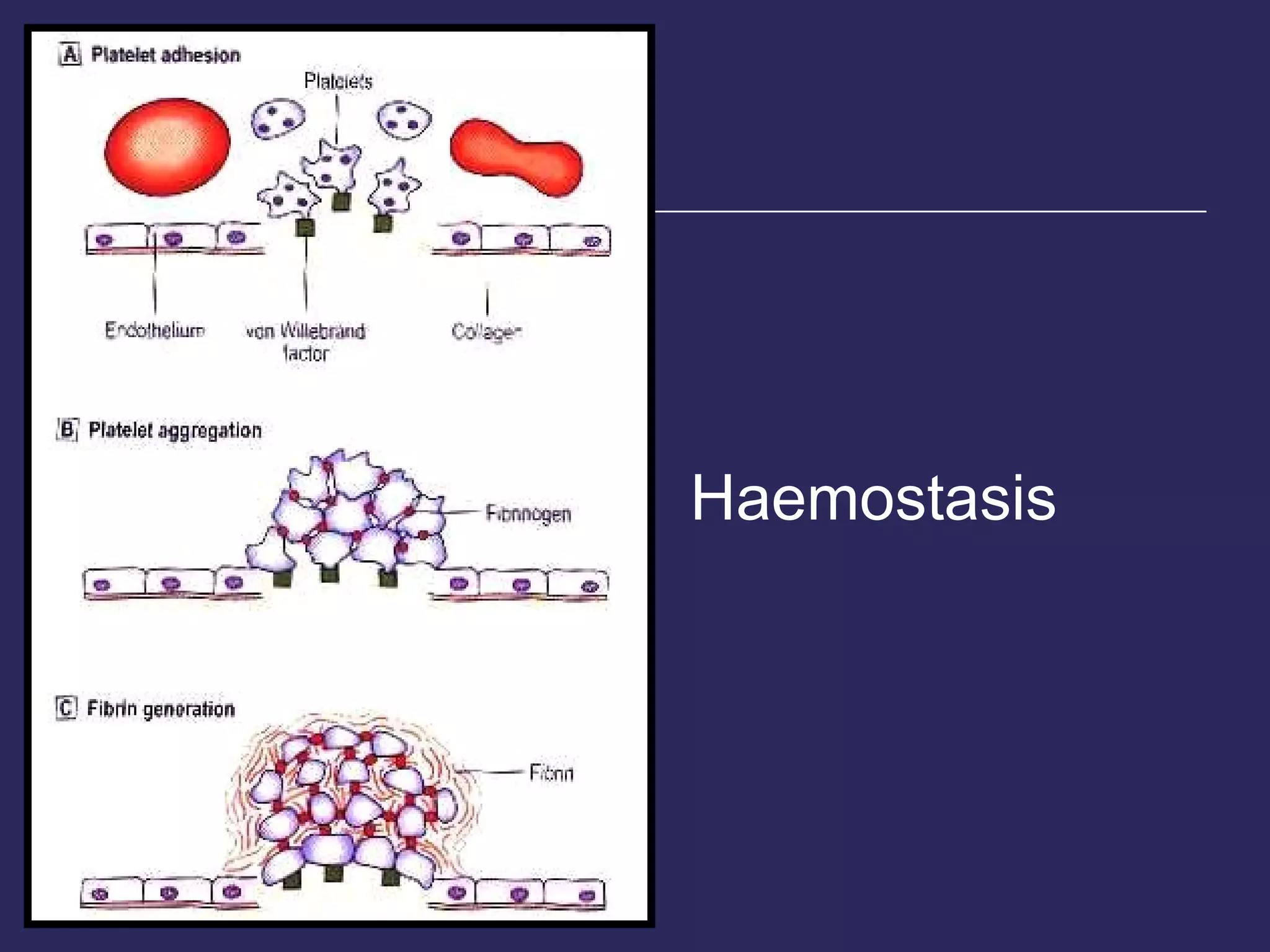
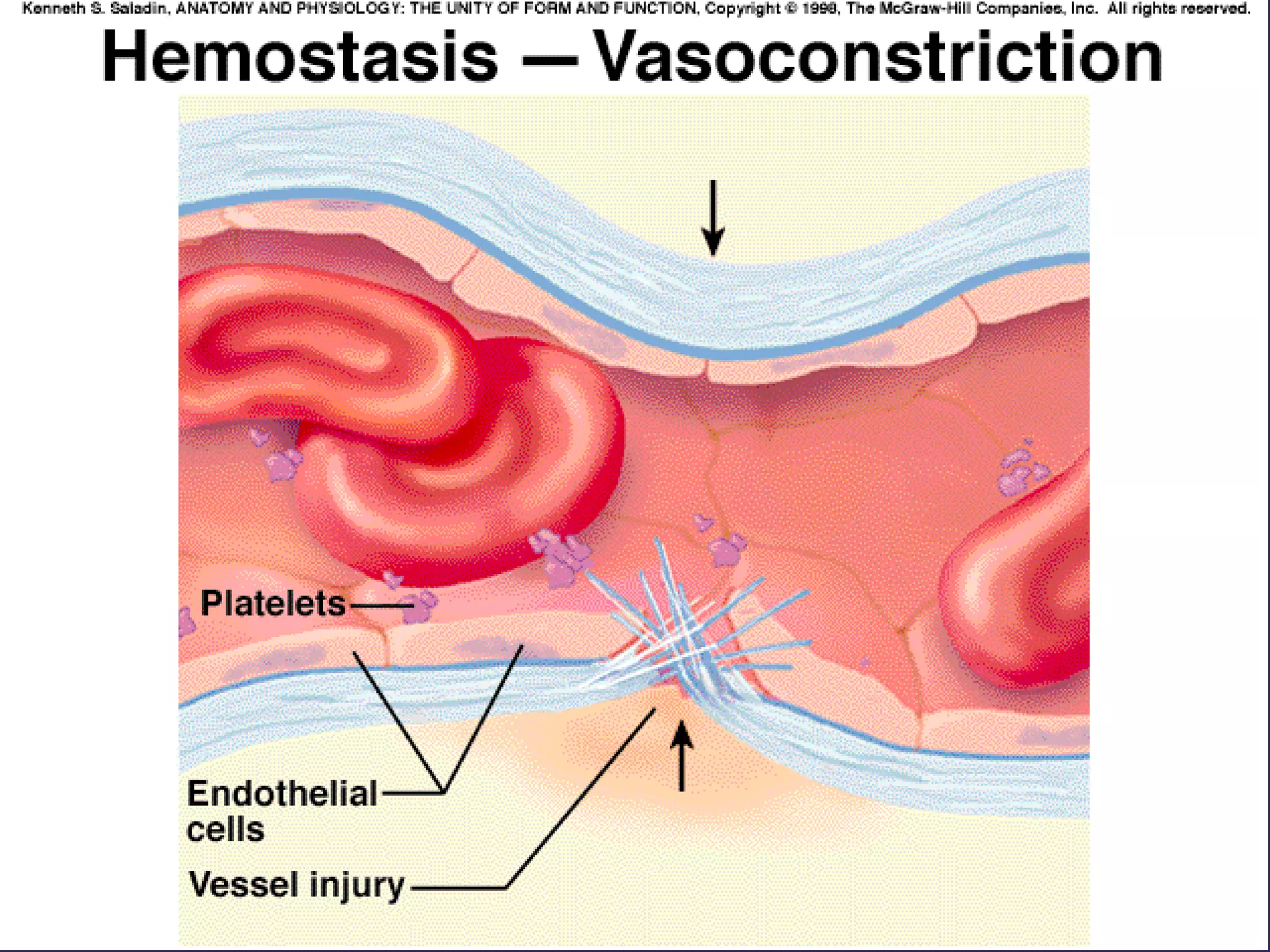
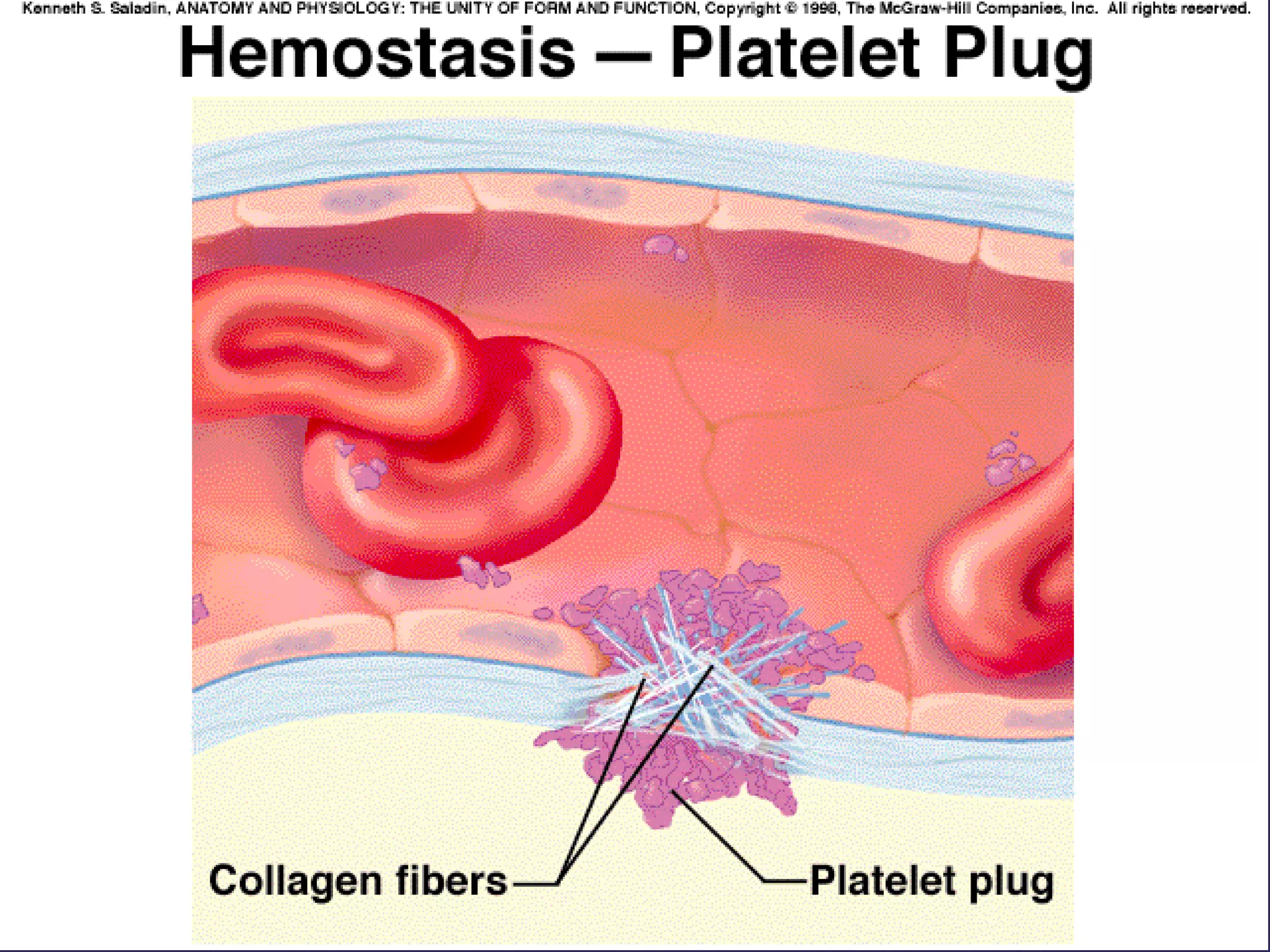
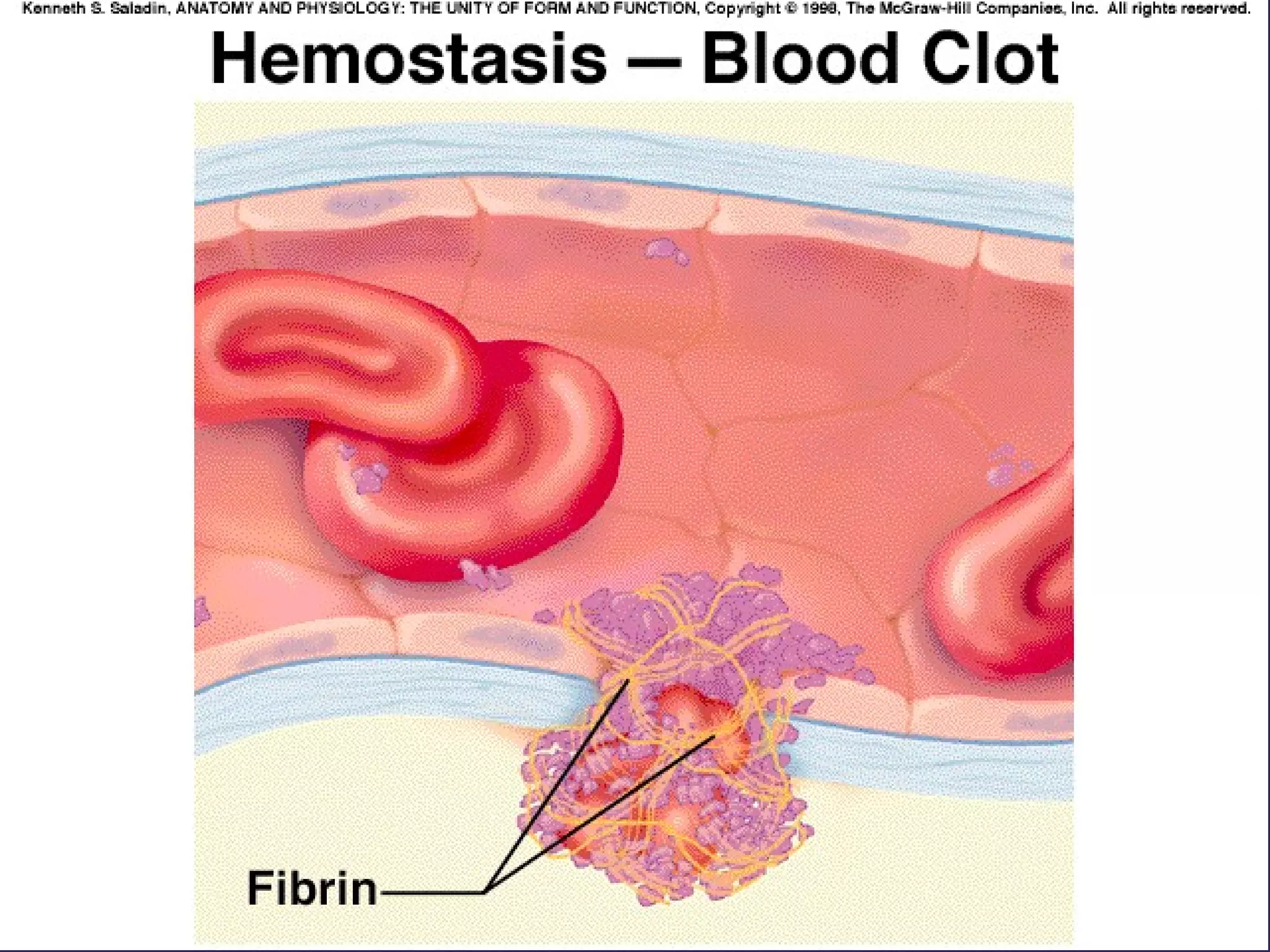
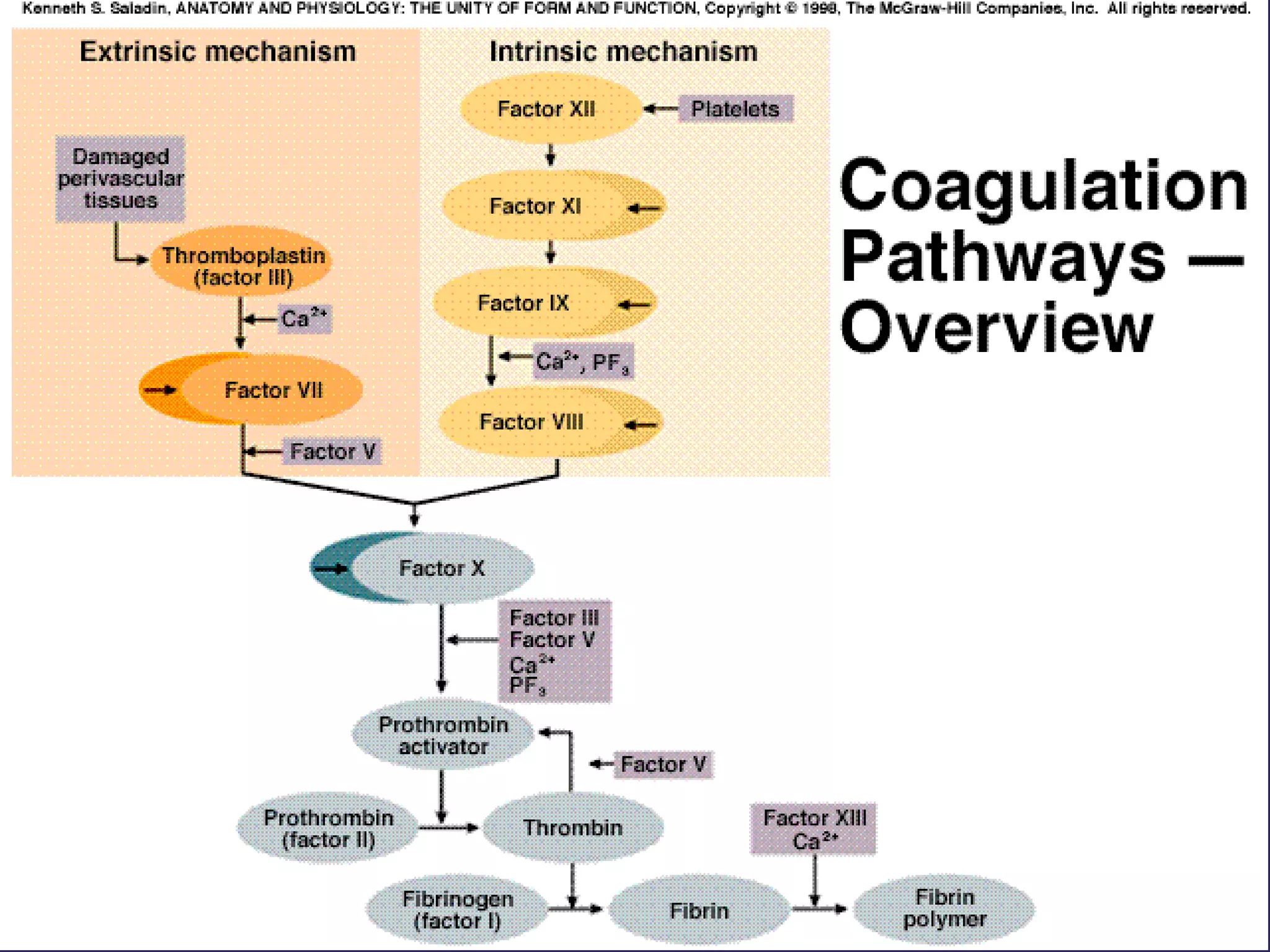
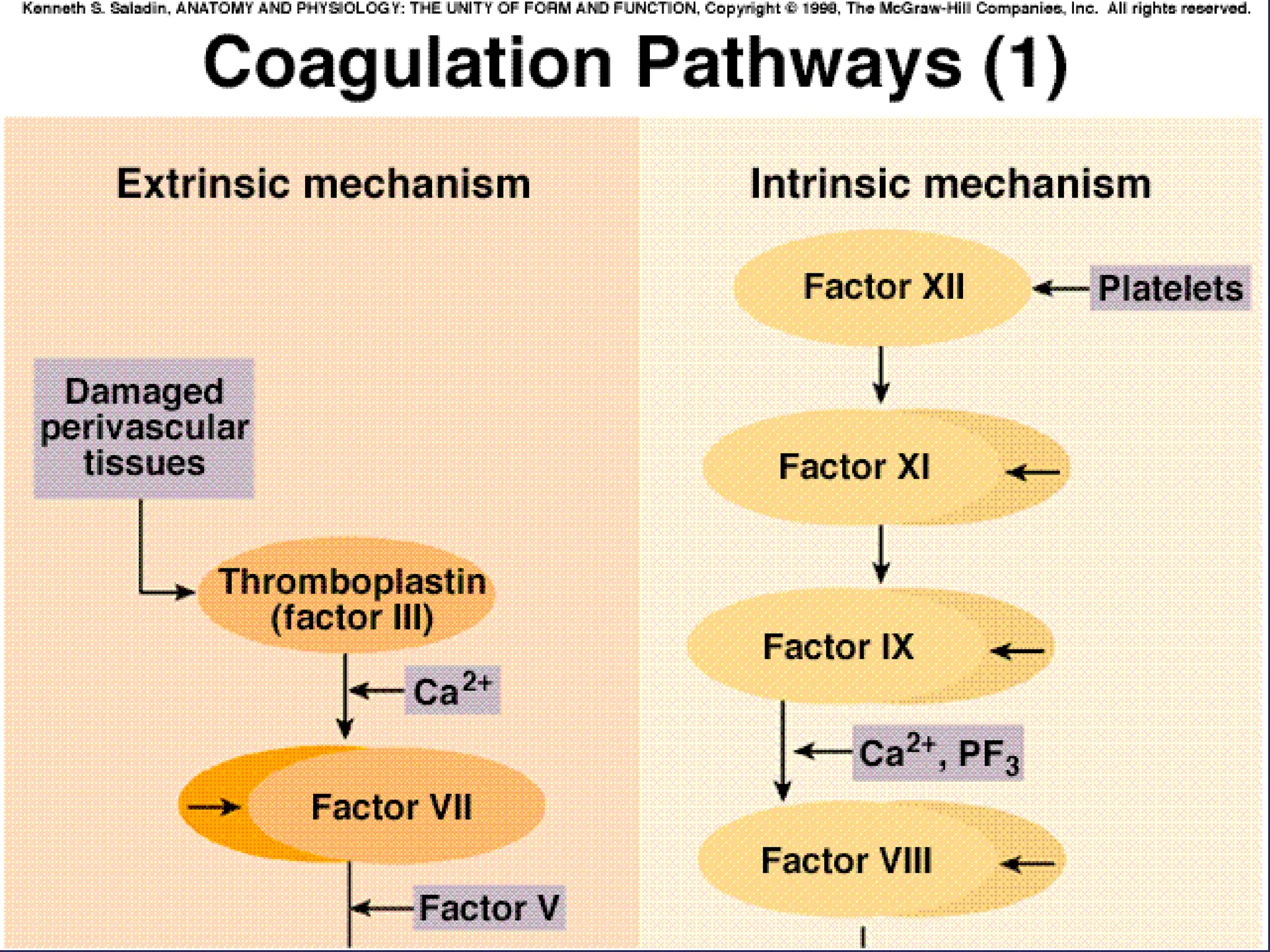
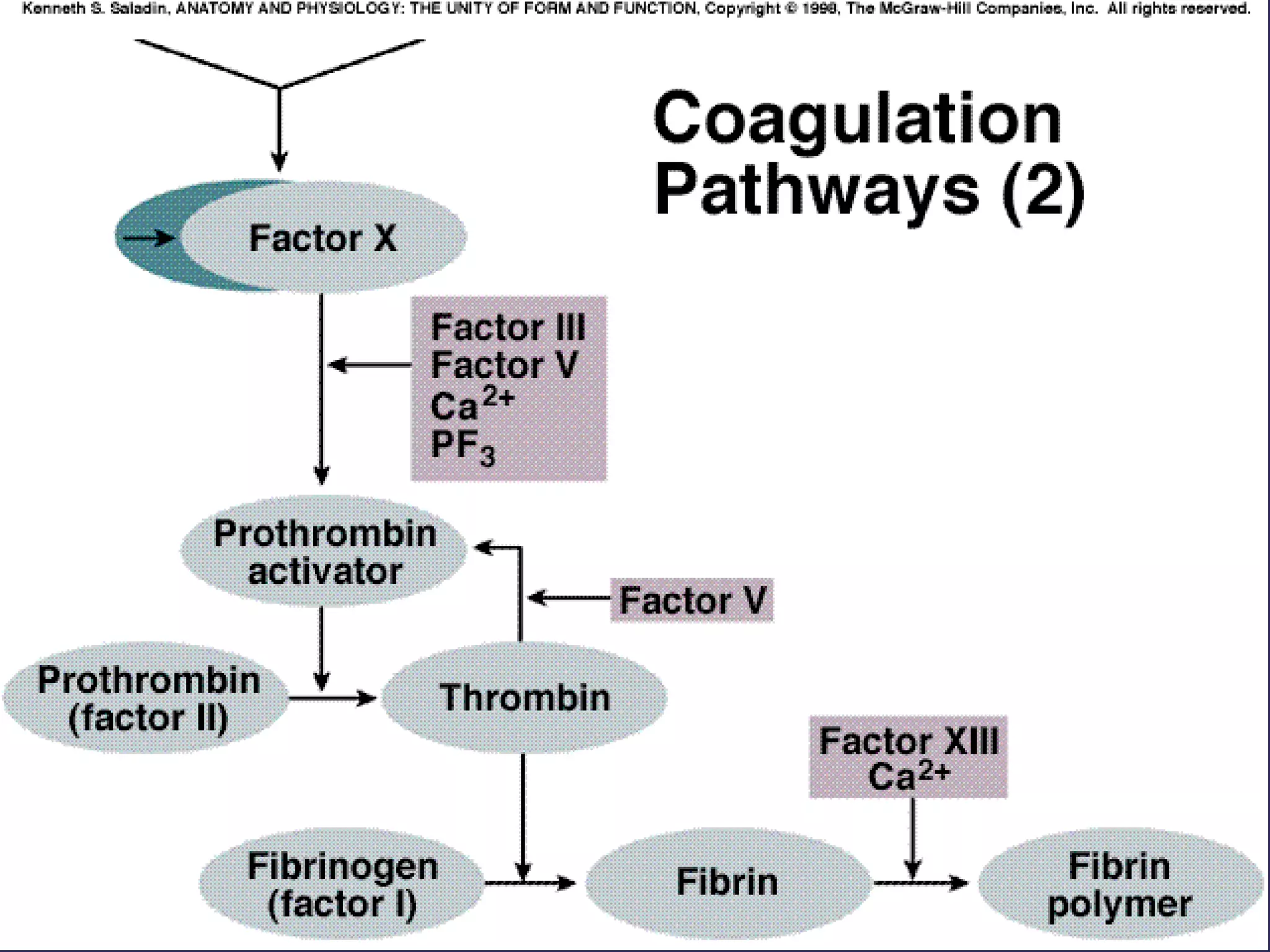
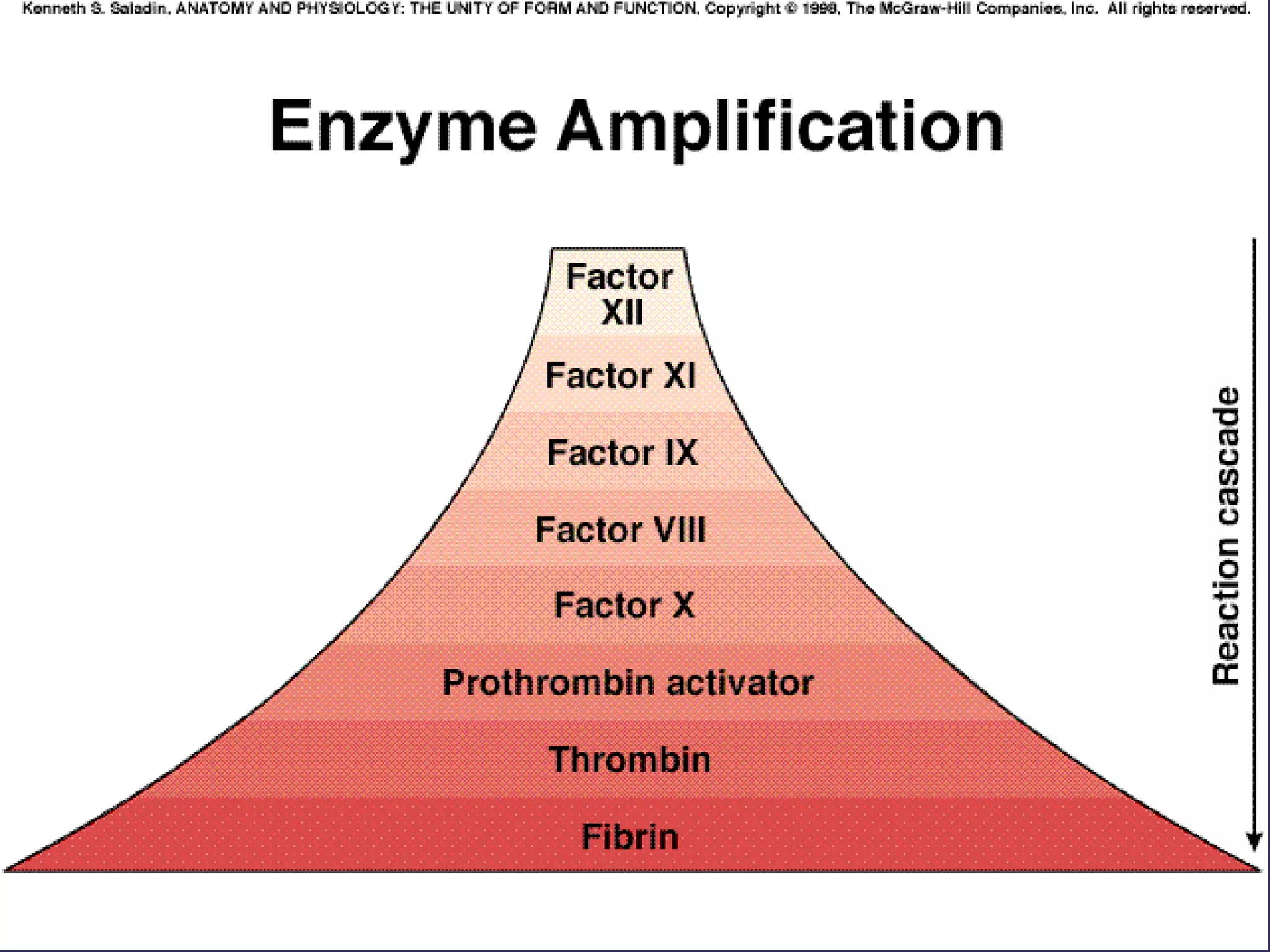
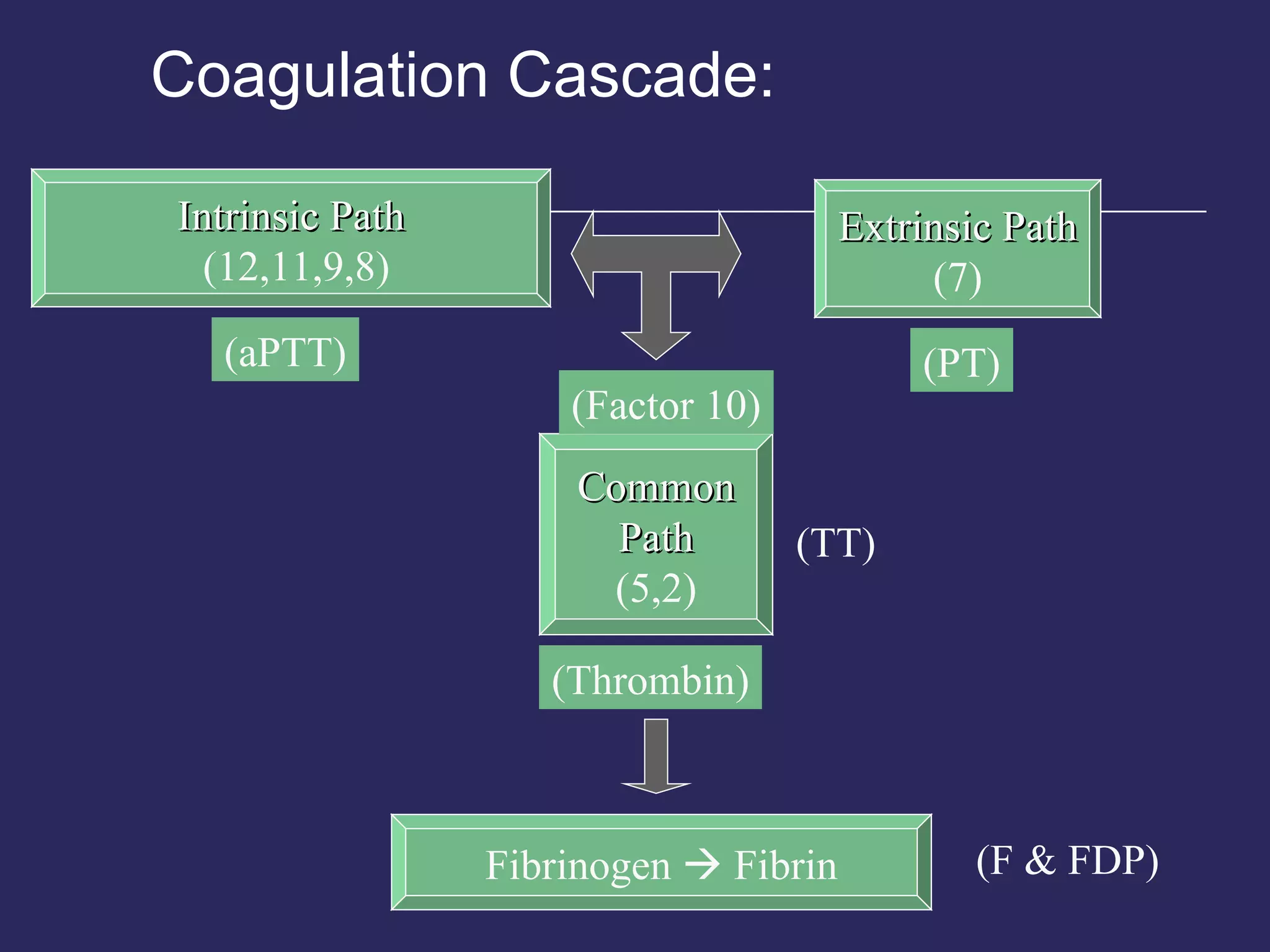
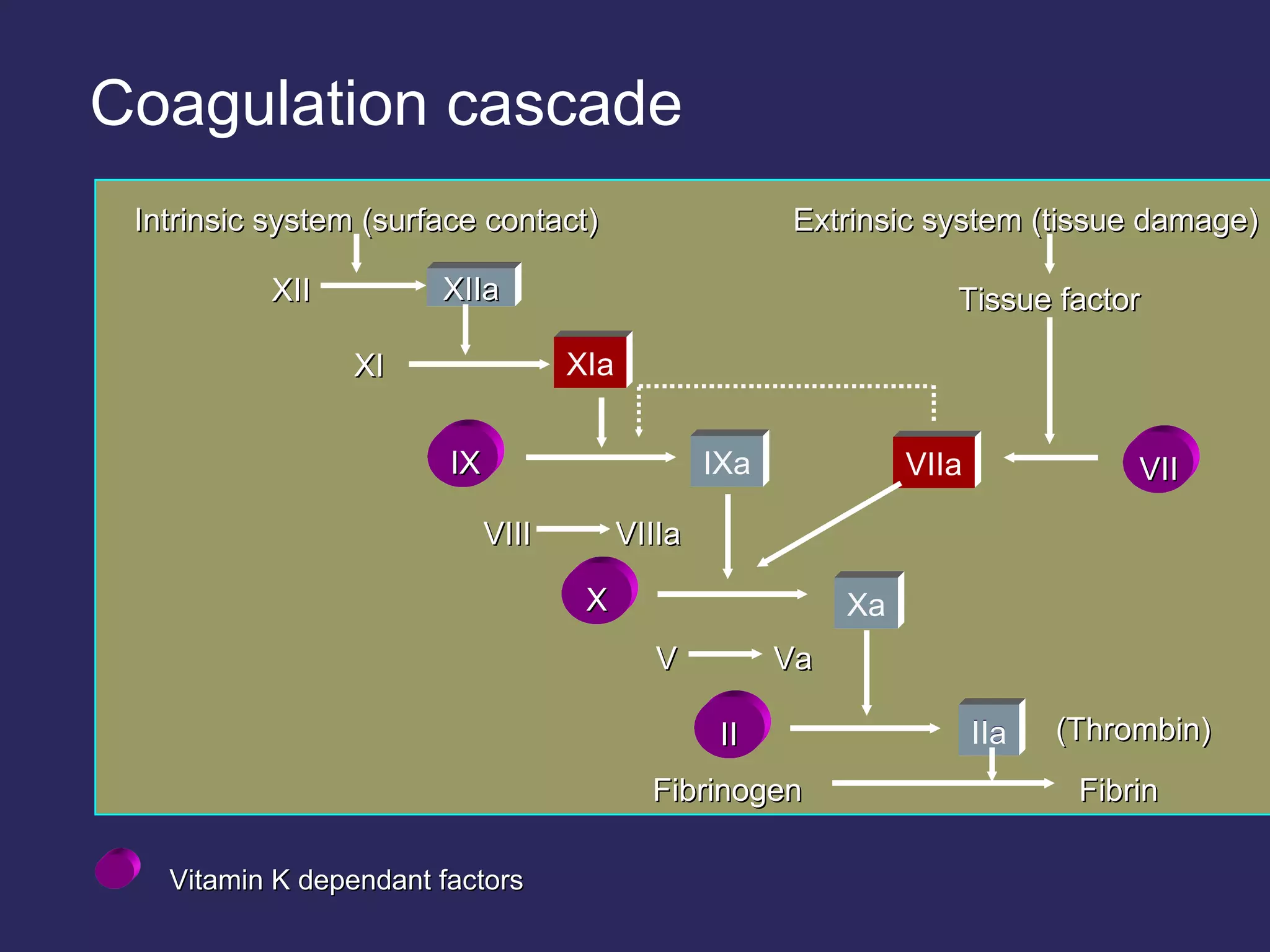
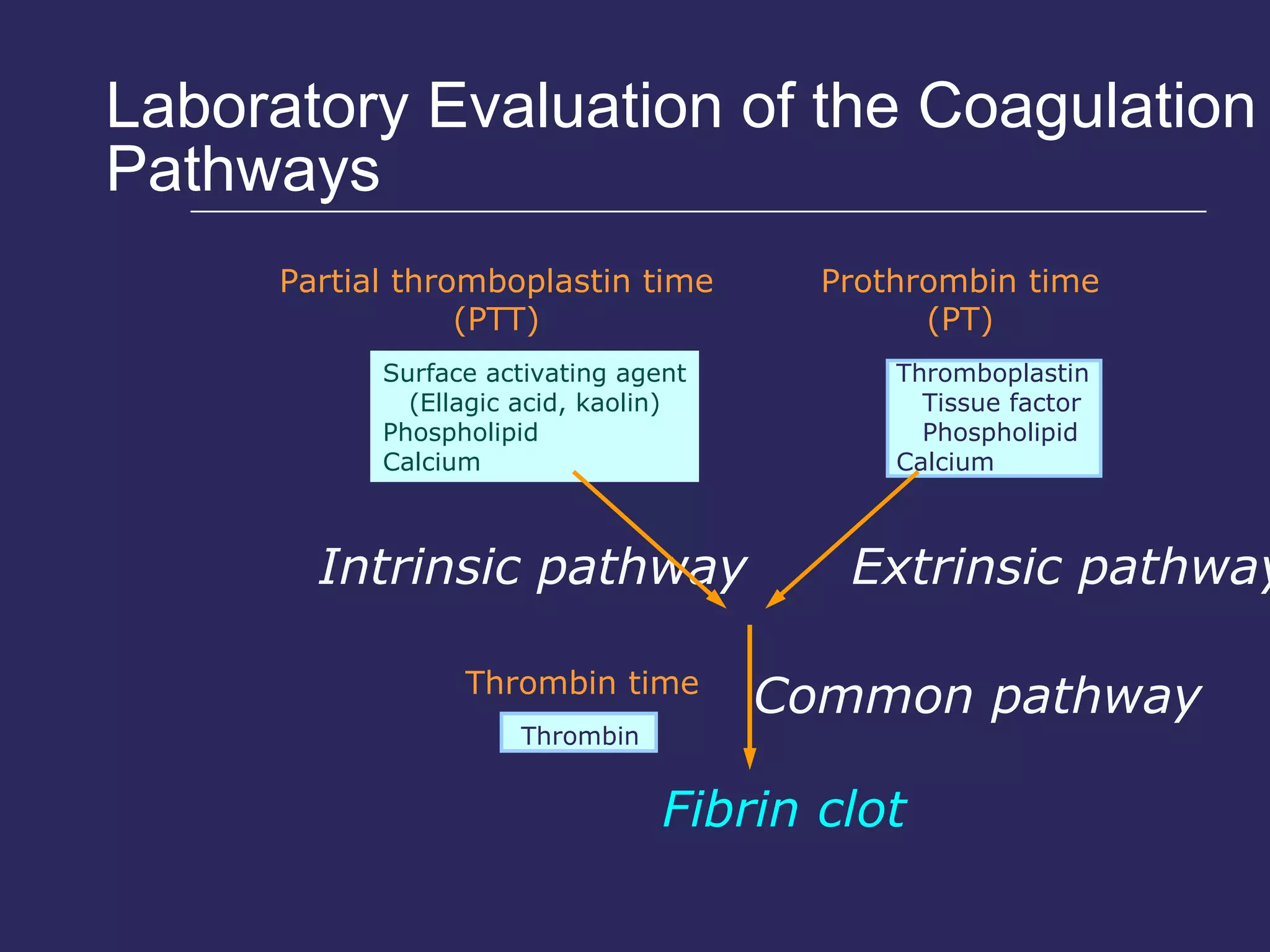
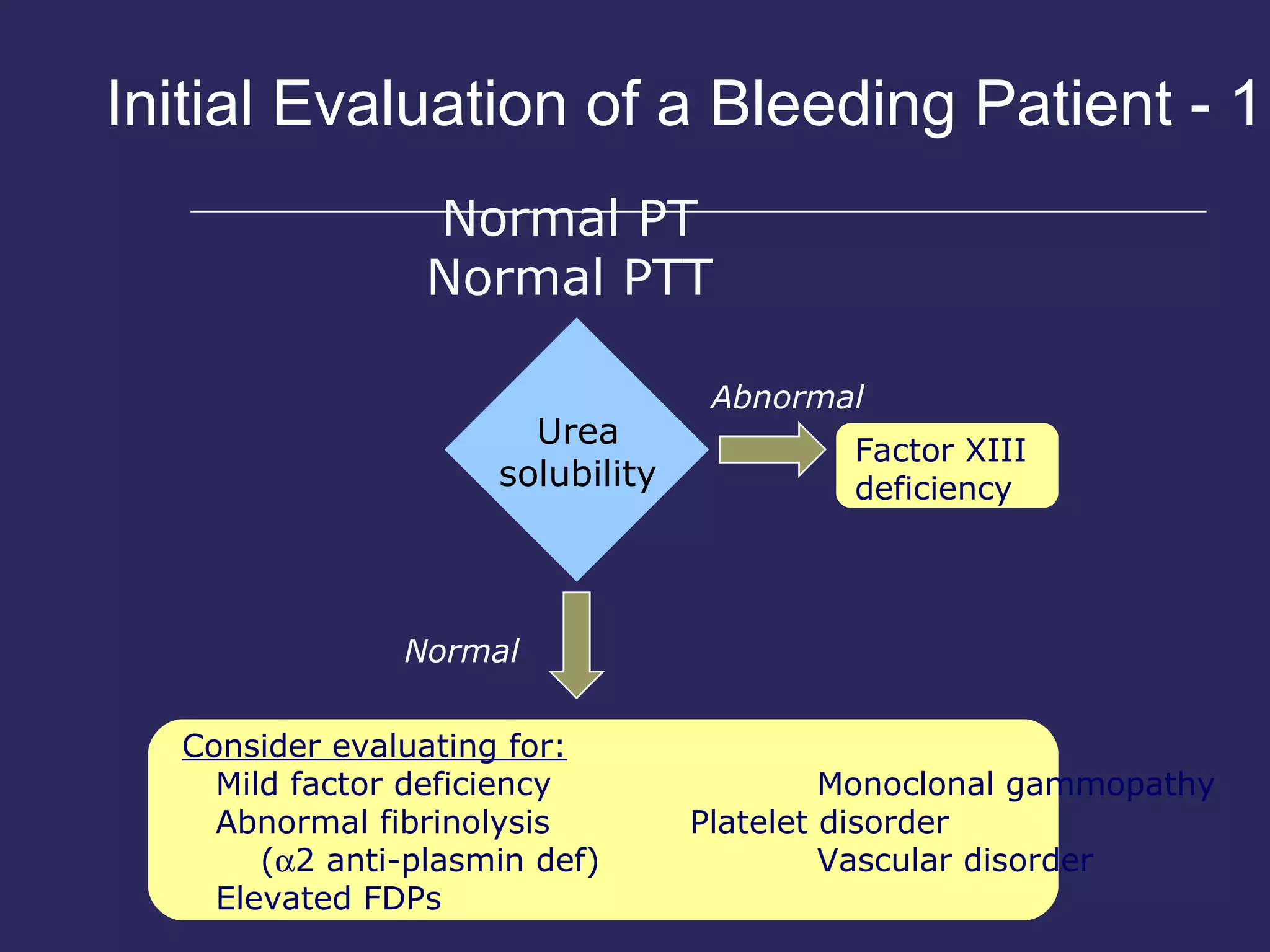
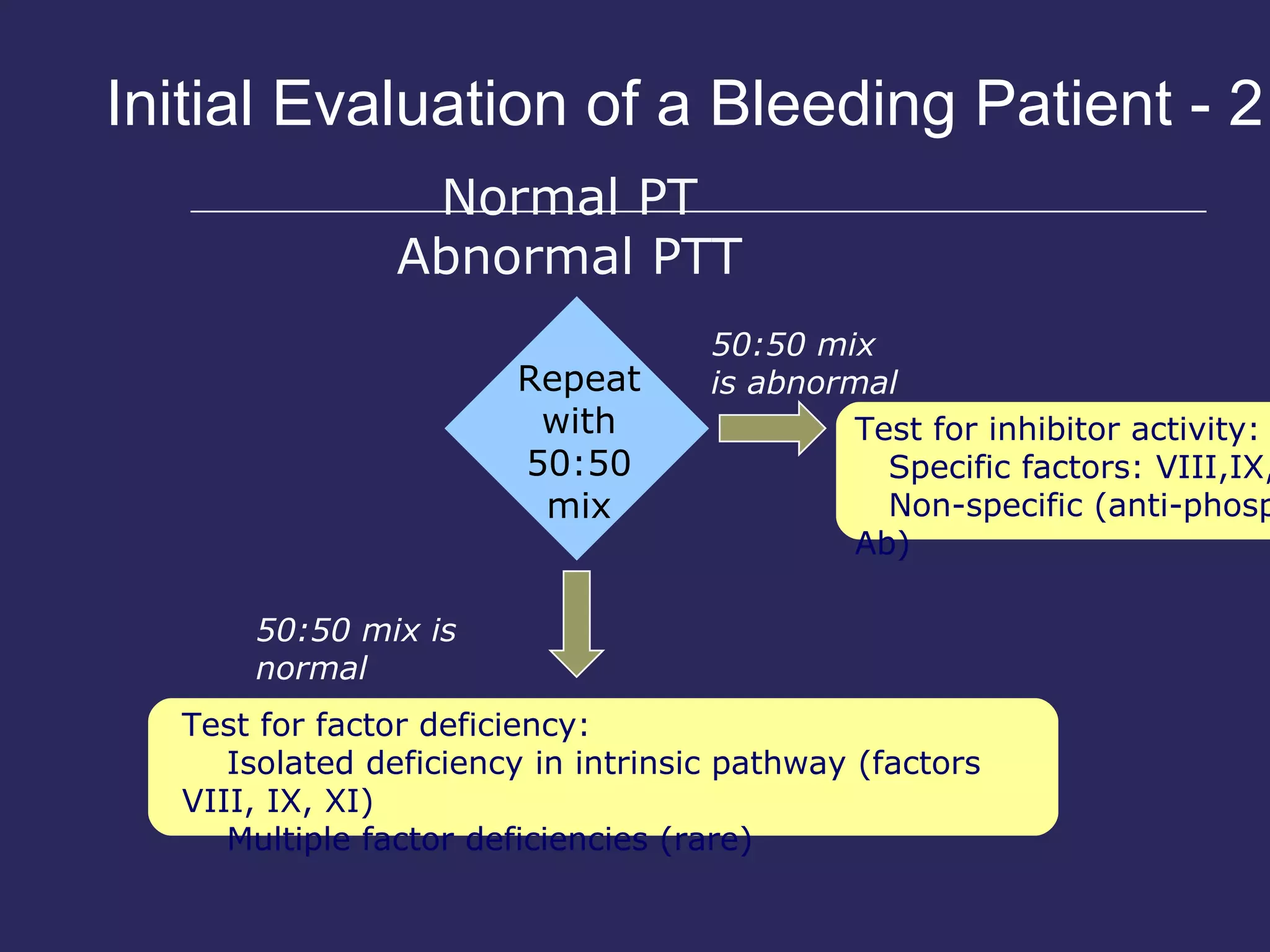
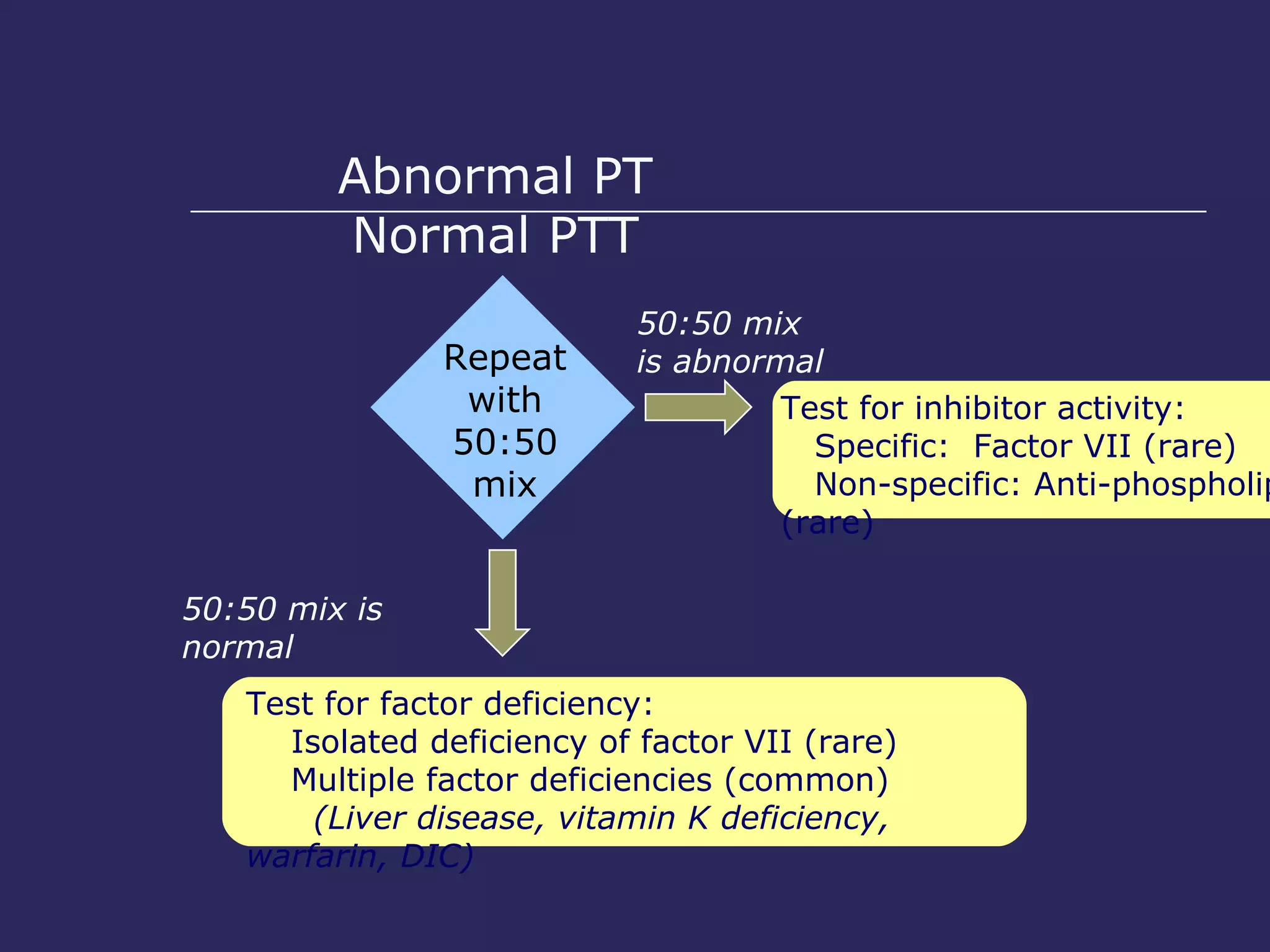
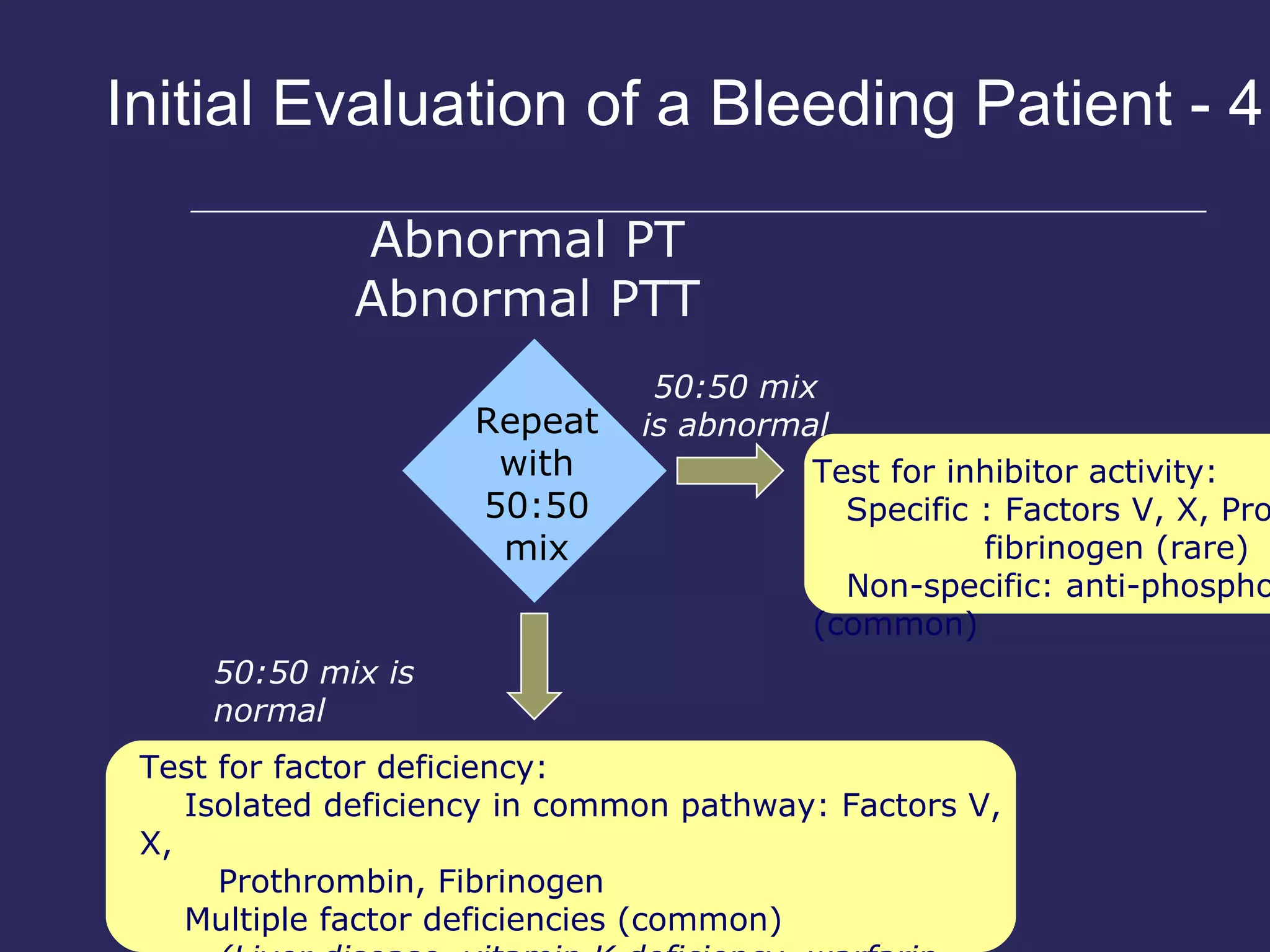
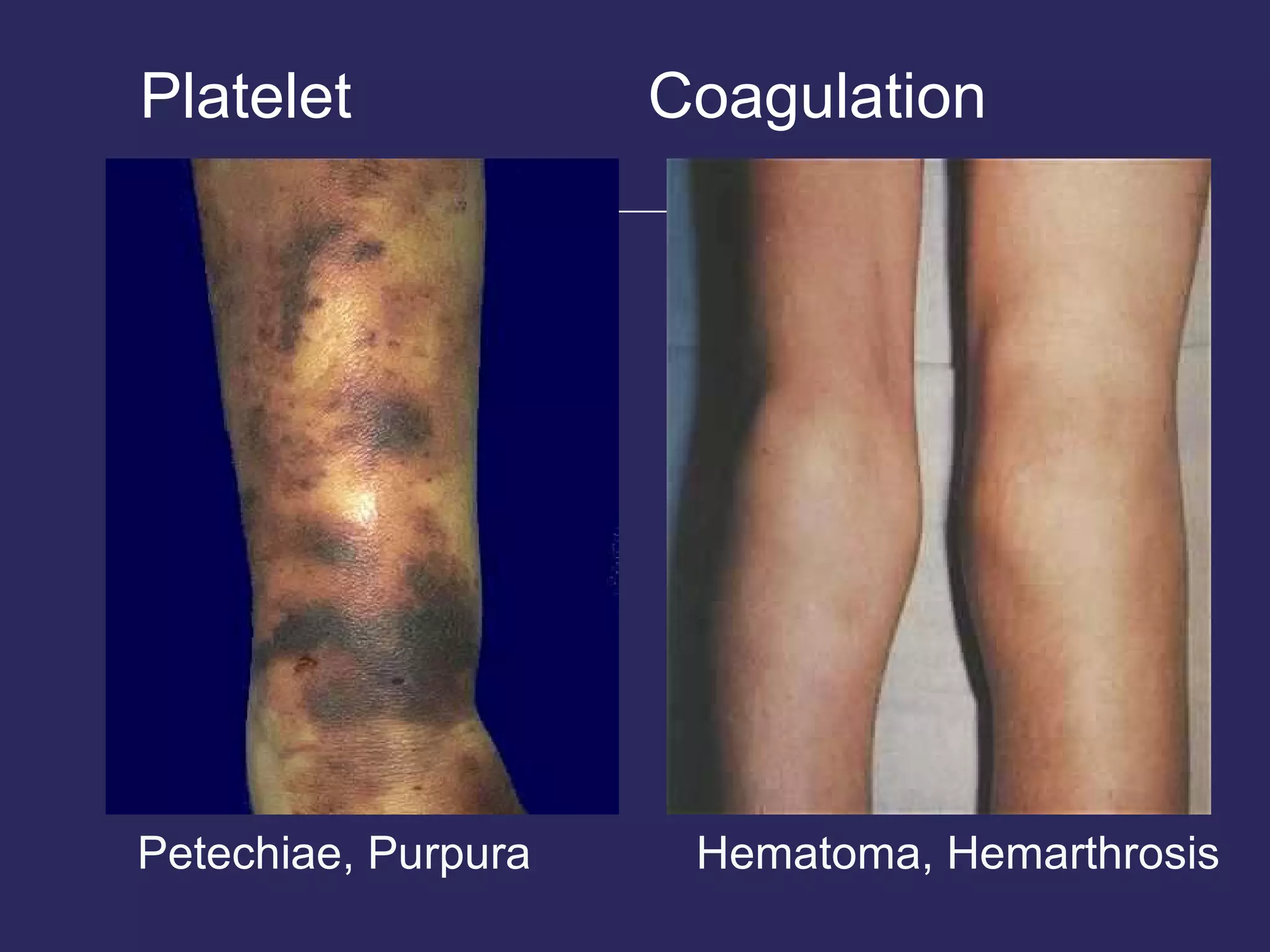
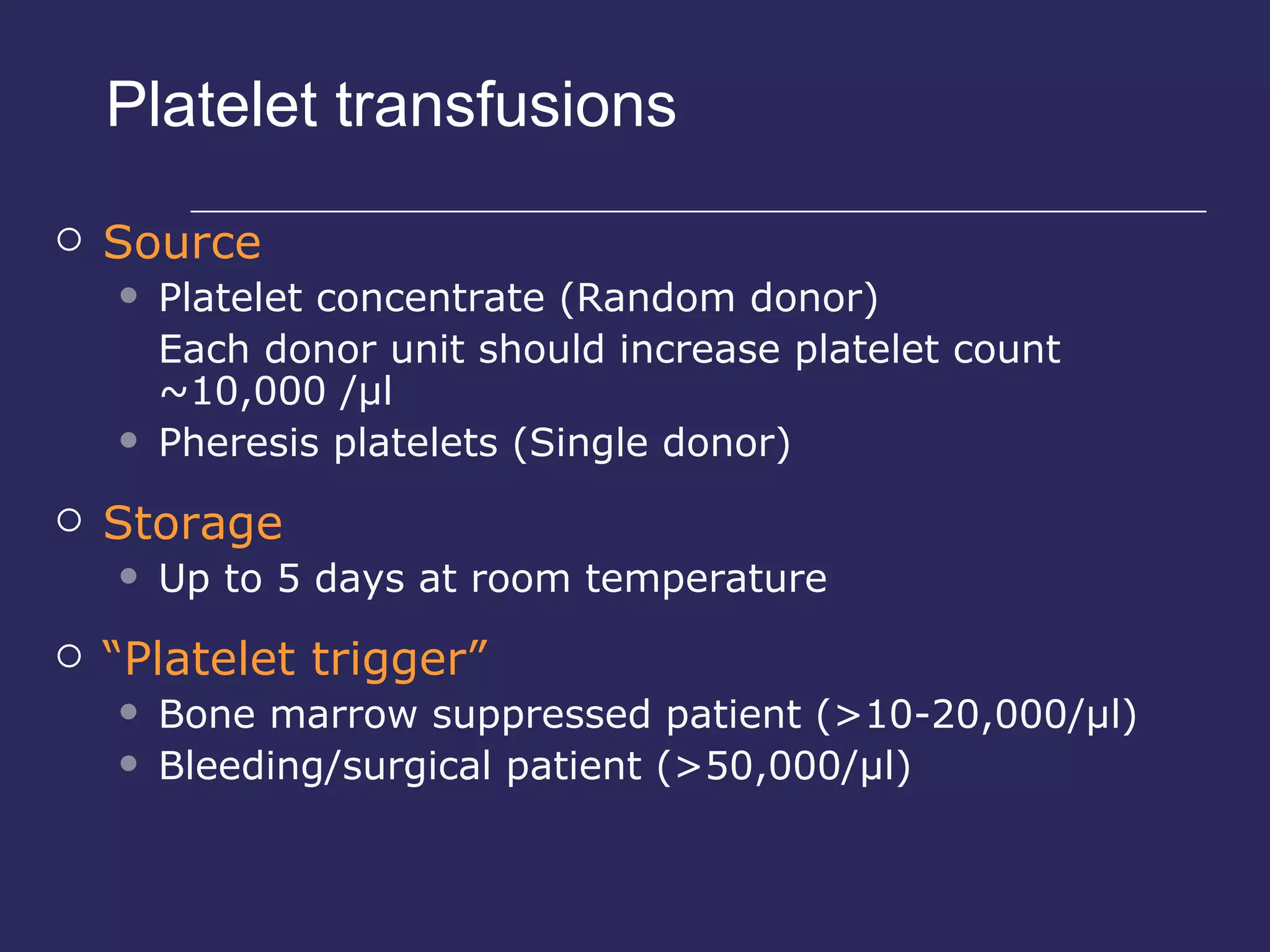
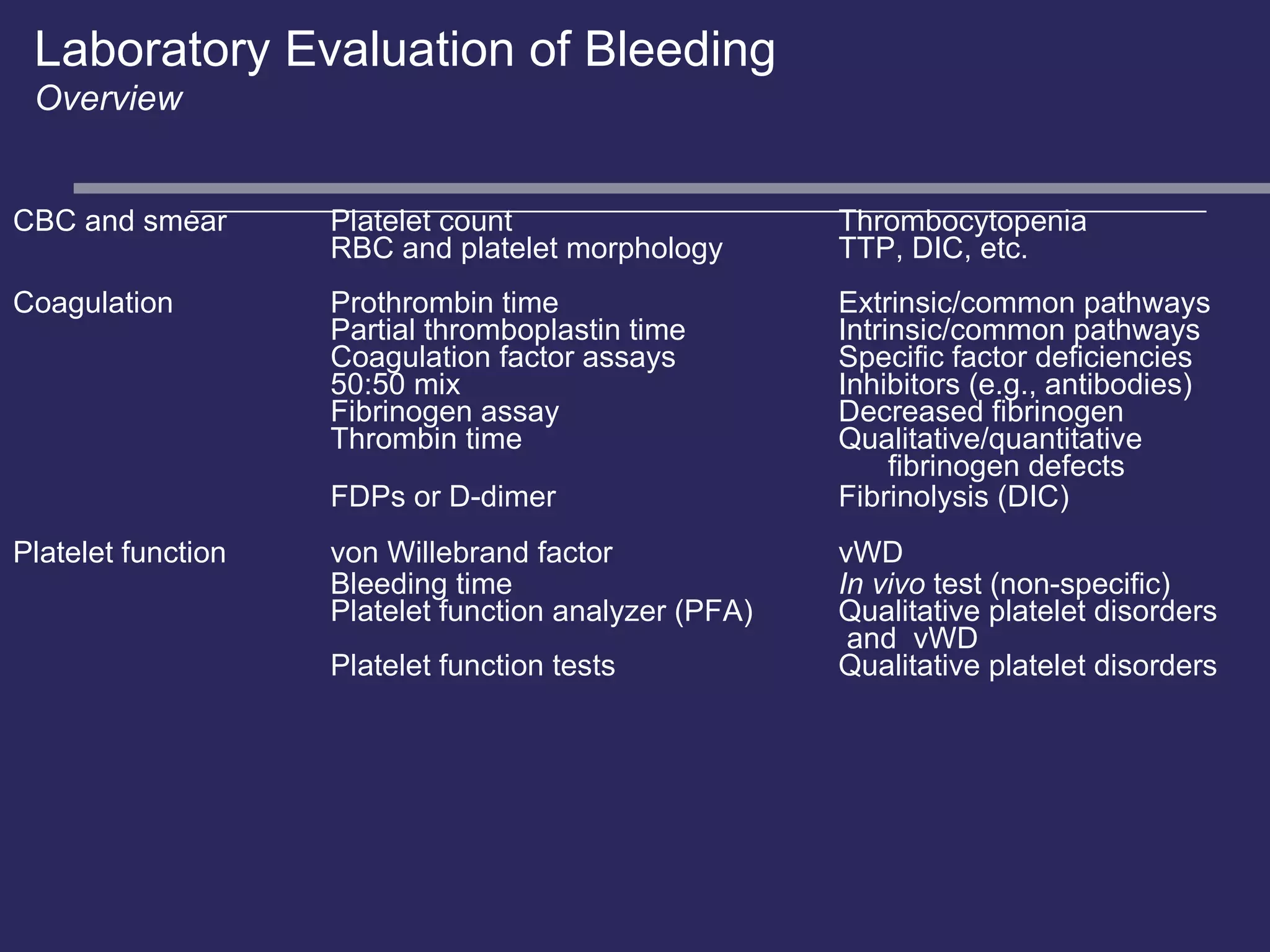
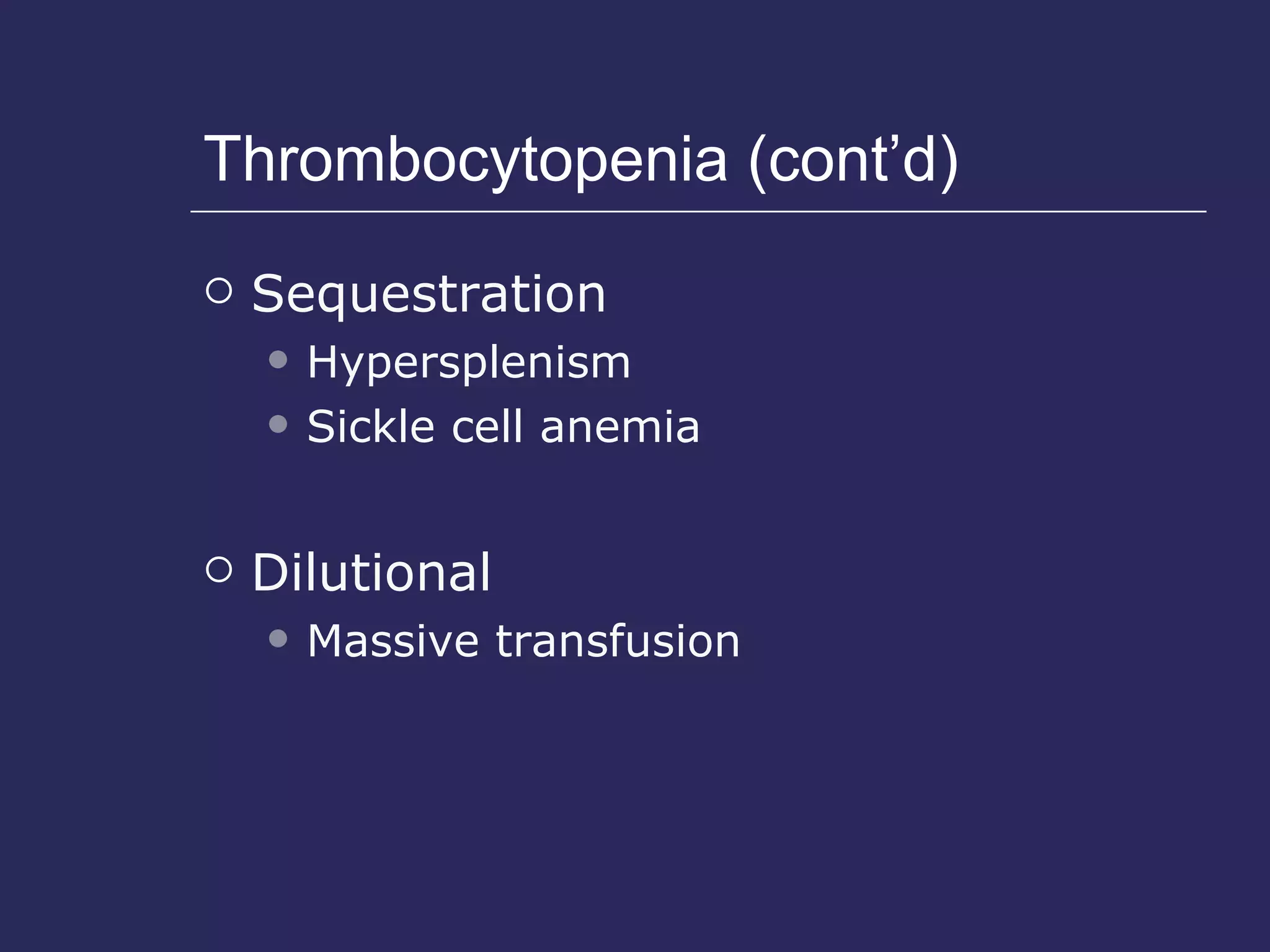
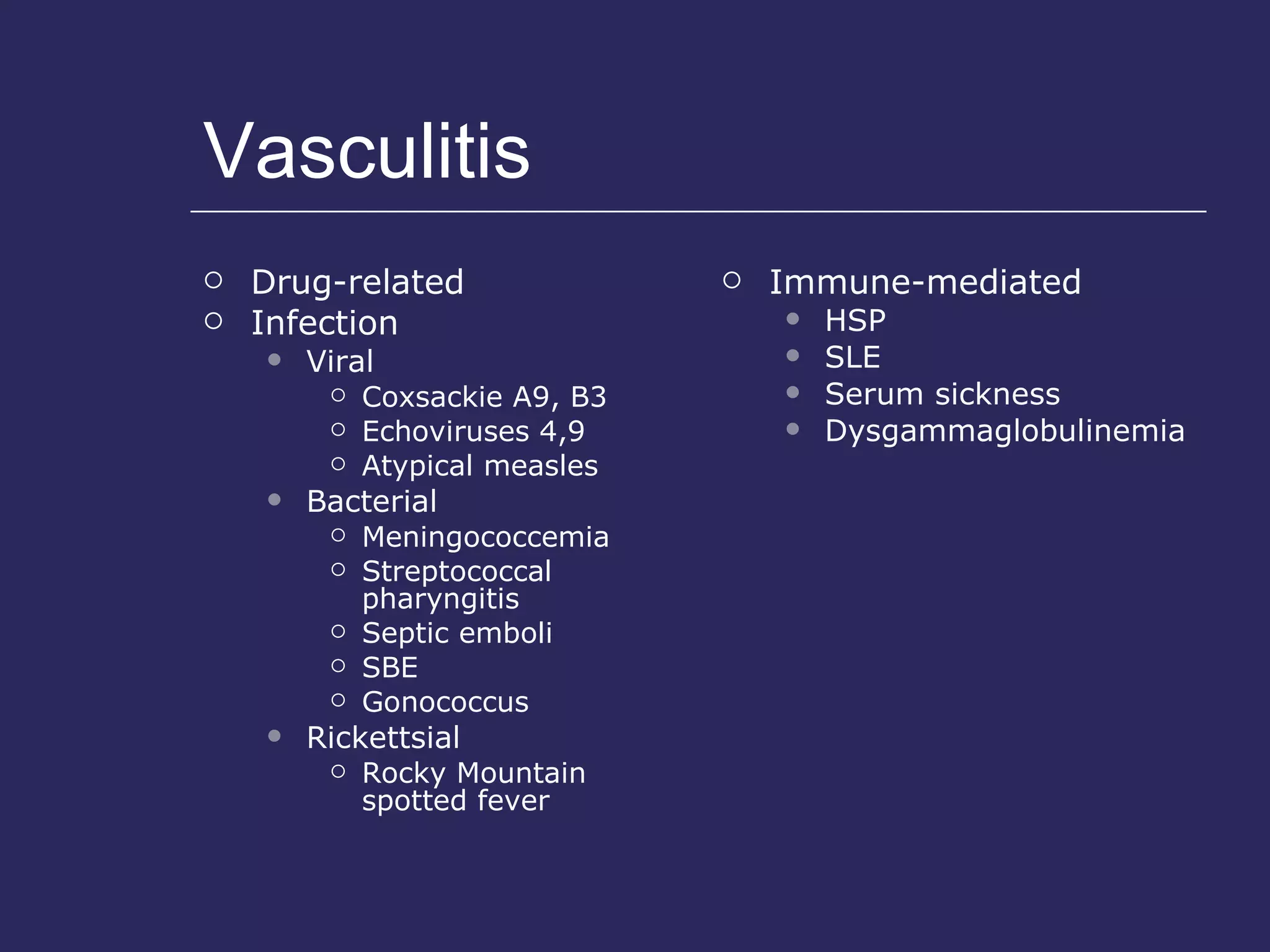
The document discusses various bleeding disorders including their causes, presentations, and treatments. It covers platelet disorders, coagulation factor deficiencies, von Willebrand disease, hemophilia A and B, disseminated intravascular coagulation (DIC), and liver disease. Treatment options discussed include cryoprecipitate, factor concentrates, DDAVP, vitamin K, fresh frozen plasma, and recombinant factor VIIa.

































































































































































































































![Apporach to lung biopsy [Auto-saved].pptx latest](https://cdn.slidesharecdn.com/ss_thumbnails/apporachtolungbiopsyauto-saved-251211225655-93258539-thumbnail.jpg?width=640&height=640&fit=bounds)



