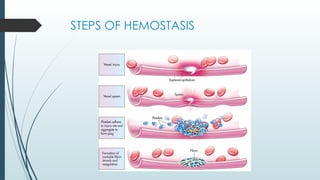
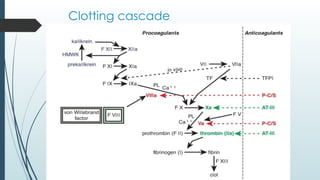
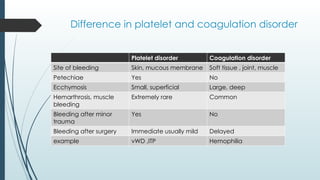
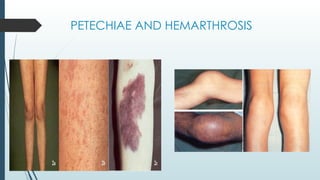
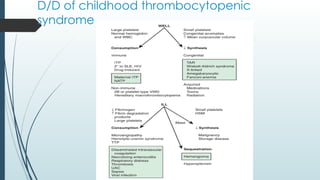
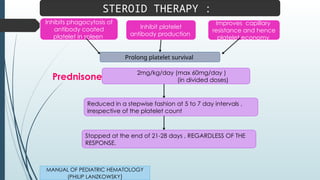
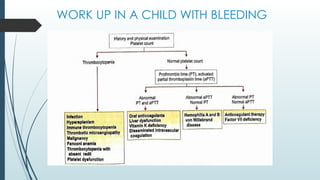
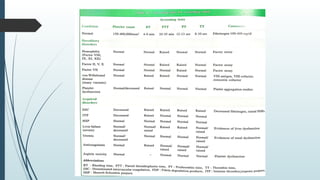
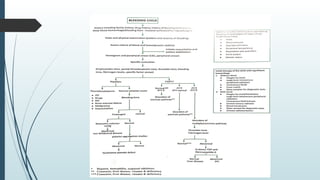
The document discusses bleeding and platelet disorders, detailing the hemostatic process and the differences between platelet and coagulation disorders. It covers various conditions such as hemophilia, von Willebrand disease, and Immune Thrombocytopenic Purpura (ITP), including their symptoms, diagnosis, and treatment options. Additionally, it highlights the importance of clinical evaluations and laboratory tests in managing these disorders.