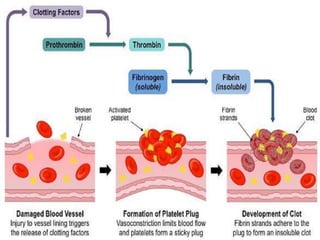
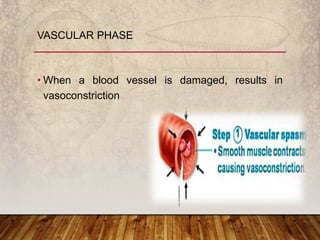
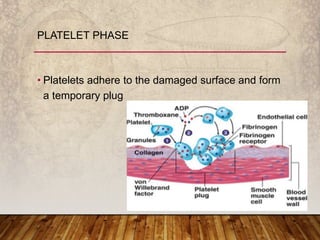
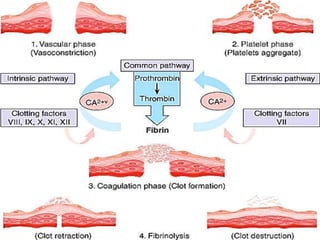
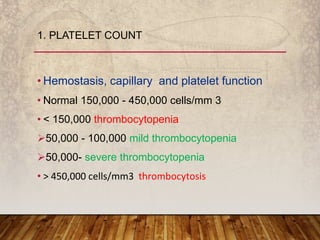
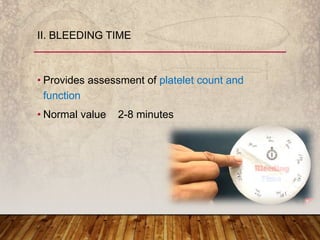
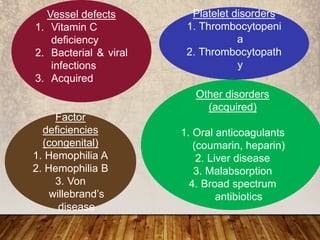
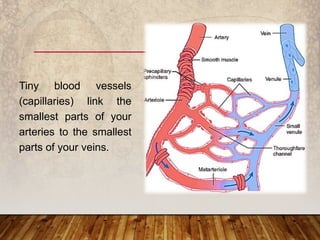
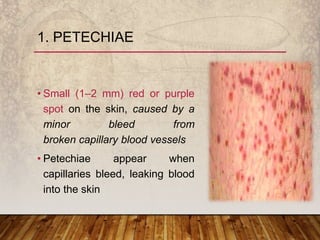
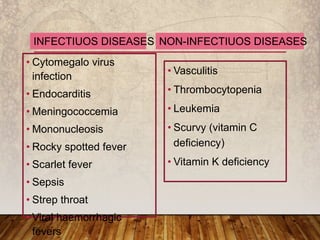
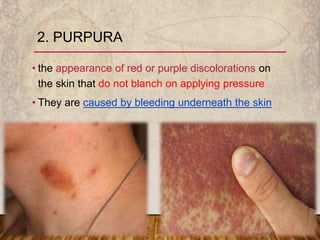
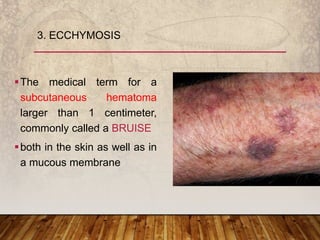
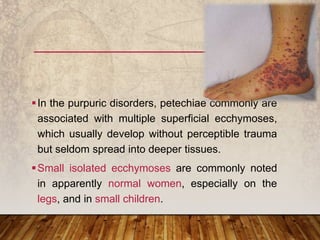
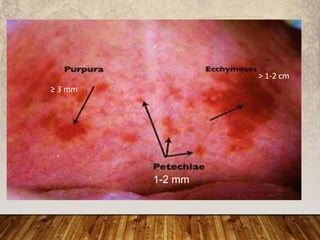
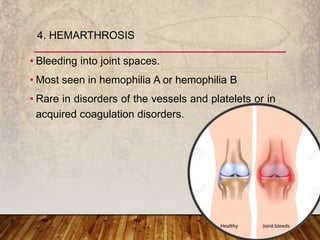
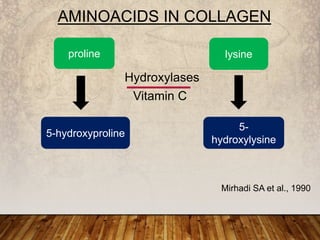
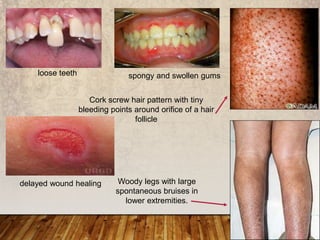
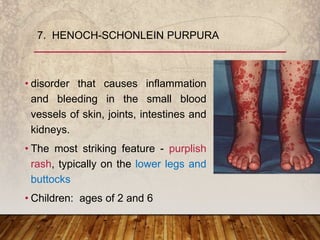
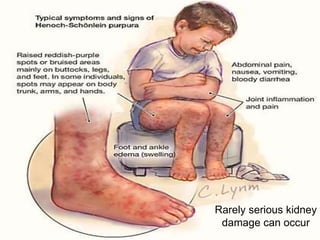
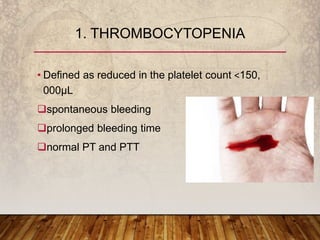
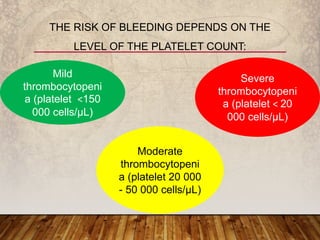
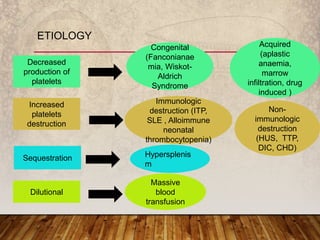
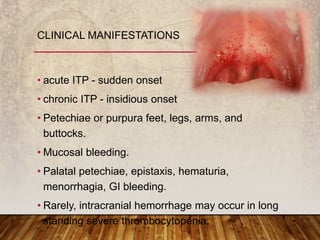
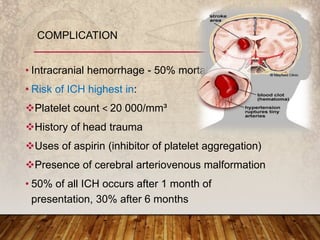
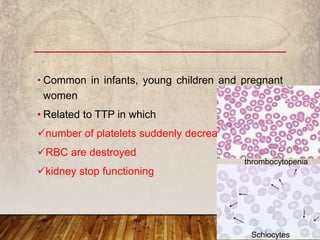
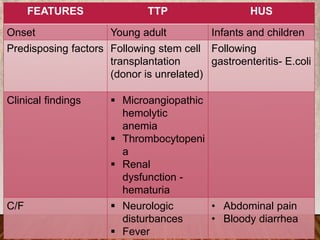
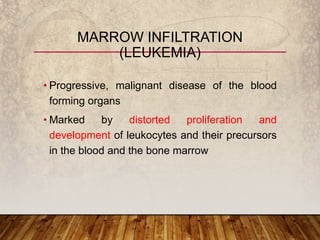
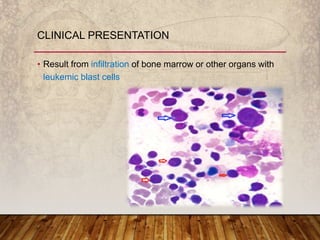
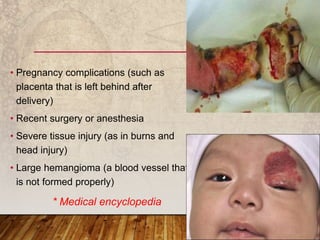
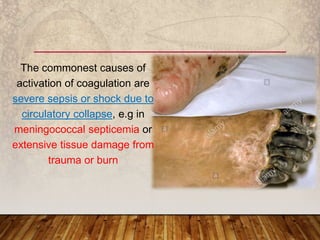
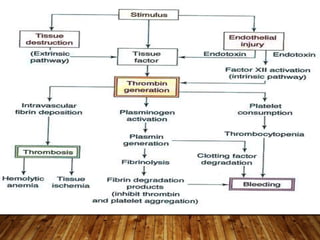
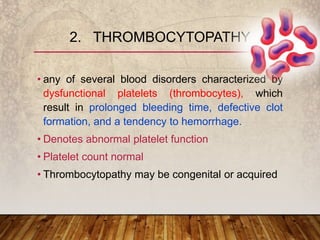
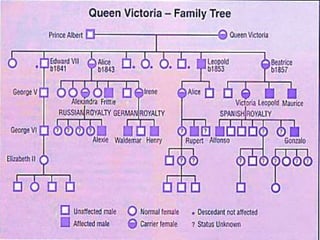
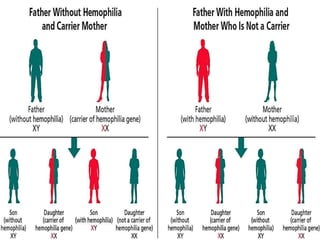
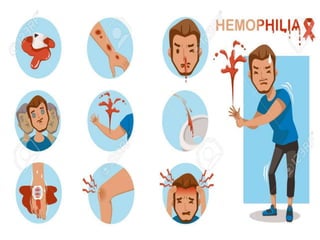
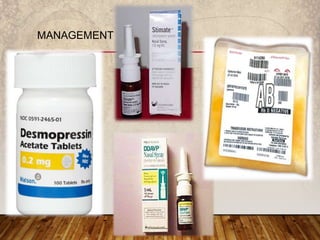
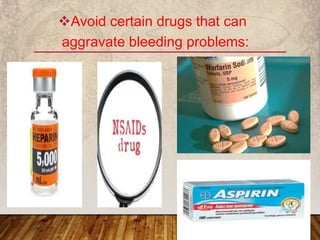
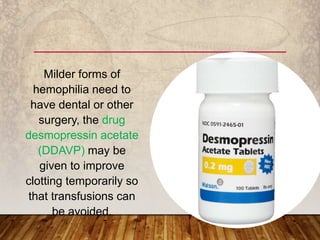
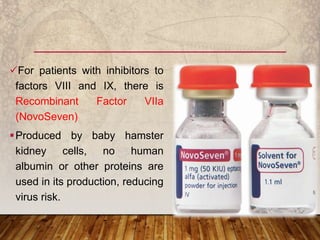
This document provides an overview of bleeding disorders, their causes, and management. It discusses how bleeding disorders are caused by abnormalities in hemostasis and coagulation that result in spontaneous or easy bruising. Various types of bleeding disorders are described, including those caused by platelet disorders like thrombocytopenia or vascular defects like vitamin C deficiency. Screening tests for bleeding disorders like platelet count, bleeding time, and INR are also outlined. Effective management of bleeding disorders depends on accurately identifying the underlying cause.